





| 图片: | |
|---|---|
| 名称: | |
| 描述: | |
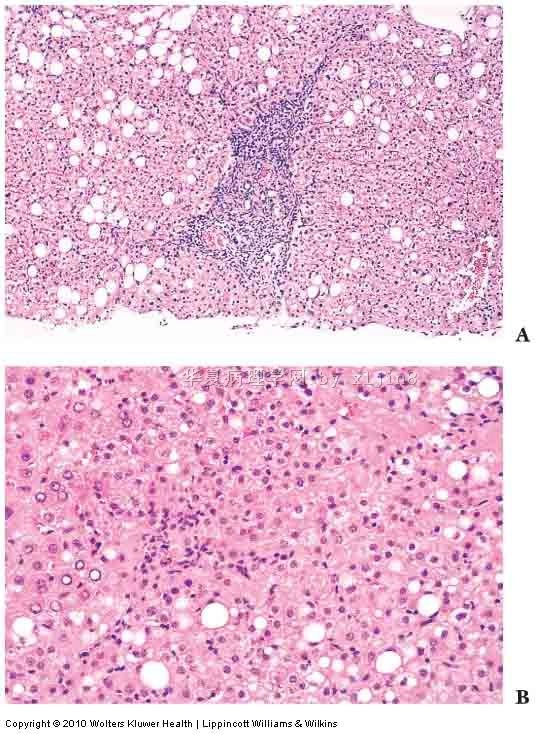
- 男/58岁,英国人,肝组织活检。
-
本帖最后由 于 2010-02-18 08:55:00 编辑
非常感谢望月老师非常详细的对“肝豆状核变性”病理学进行了复习和讲解,使我们增加了新的知识。
我想病理讨论的目的就在于让我们能独立的思考问题、查阅文献资料、反复的推敲诊断和鉴别诊断、充分的发挥已有实验室条件、密切的联系临床。即使是诊断上有偏差,也毫无疑问!
此外,即使诊断有困难,也可提出大致的诊断意见,建议到上级医院进行的检查。对上级医院的诊断意见也要进行分析,总结自己在什么方面可进一步提高。千万不能认为上级医院的诊断一定就是正确的,还是要加以分析。并对会诊医生要有所了解。因为,即使是上级医院,但会诊医生有可能不具有此专业特长。
本病例无肝窦状核变性的家族史;临床实验室有关铜代谢的生化检查均正常;也无神经系统改变。
请大家再为这位英国病人想象办法,谢谢!

- xljin8
-
本帖最后由 于 2010-02-11 12:15:00 编辑
鉴别诊断:
1、胆酸盐淤积:汇管区周围肝细胞肿胀、苍白、粗颗粒状(含有铜结合蛋白),亦可见Mallory小体,晚期见胆红素包涵体。CK7染色汇管区细胞(+),向小叶中心渐减少。
2、 酒精性肝损伤、肝硬变:早期小叶中央脂肪变,融合大脂肪,气球样肝细胞伴或不伴Mallory小体,细胞核空泡变,肝细胞坏死凋亡,炎细胞浸润,中性粒为主,“鸡爪样纤维化”(特征性),局部静脉纤维化、硬化
3、慢性淤胆性肝硬变
4、 印度儿童肝硬变
- 广州金域病理
-
本帖最后由 于 2010-02-11 12:12:00 编辑
肝豆状核变性Wilson’s disease
病理特点:
肝脂肪变性,有时脂性肉芽肿
气球样变肝细胞、糖原化核(核内空泡)
汇管区肝细胞内铜沉积(褐色颗粒状)
汇管区周围肝细胞内Mallory小体
汇管区淋巴细胞等浸润
肝细胞坏死,间质进行性纤维化,肝小叶再生,肝硬化。
组织化学染色:
Timm银染色,最敏感
Rhodanine染色
地衣红染色
以上染色后胞浆内见红色颗粒。
注意:无症状的患者,因铜弥漫分布于胞浆中,不易测出,或至多弱阳性。- 广州金域病理
肝豆状核变性Wilson’s disease临床特点
本病大多在10~25岁间出现症状,男稍多于女,同胞中常有同病患者。一般病起缓渐,临床表现多种多样,主要症状为:
1、神经系统症状:常以细微的震颤、轻微的言语不清或动作缓慢为其首发症状,以后逐渐加重并相继锥体外系症状
2、肝脏症状:儿童期患者常以肝病为首发症状,成人患者可追索到“肝炎”病史。肝脏肿大,质较硬而有触痛,肝脏损害逐渐加重可出现肝硬化症状,脾脏肿大,脾功亢进,腹水,食道静脉曲张破裂及肝昏迷等。
3、角膜色素环:角膜边缘可见宽约2~3mm左右的棕黄或绿褐色色素环,用裂隙灯检查可见细微的色素颗粒沉积,为本病重要体征,一般于7岁之后可见。
4、肾脏损害:因肾小管尤其是近端肾小管上皮细胞受损,可出现蛋白尿、糖尿、氨基酸尿、尿酸尿及肾性佝偻病等。
5、溶血。可与其它症状同时存在或单独发生,由于铜向血液内释放过多损伤红细胞而发生溶血。
- 广州金域病理
昨晚查阅一些资料,将Wilson病总结如下:
肝豆状核变性Wilson’s disease
肝豆状核变性(hepatolenticular degeneration,HLD)又称威尔逊氏病,常染色体隐性遗传的铜代谢障碍疾病。 由Wilson首先报道和描述,是一种遗传性铜代谢障碍所致的肝硬化和以基底节为主的脑部变性疾病。临床上表现为进行性加重的椎体外系症状、肝硬化、精神症状、肾功能损害及角膜色素环K-F环。
病因
本病铜代谢障碍的具体表现有:血清总铜量和铜蓝蛋白减少而疏松结合部分的铜量增多,肝脏排泄铜到胆汁的量减少,尿铜排泄量增加,许多器官和组织中有过量的铜沉积尤以肝、脑、角膜、肾等处为明显。过度沉积的铜可损害这些器官的组织结构和功能而致病。
肝豆状核变性铜代谢障碍的机制
铜作为辅基参与多种重要生物酶的合成。正常人从肠道吸收入血的铜大部分先与白蛋白疏松结合,然后进入肝细胞。在肝细胞中,铜经P型铜转运ATP酶转运到Golgi体,再与α2球蛋白牢固结合成铜蓝蛋白,然后分泌到血液中。循环中的铜90%-95%结合在铜蓝蛋白上。70%的铜蓝蛋白存在于血浆中,其余部分存在组织中。多余的铜主要以铜蓝蛋白的形式从胆汁排出体外。此病患者由于P型铜转运ATP酶缺陷,造成肝细胞不能将铜转运到Golgi体合成铜蓝蛋白,过量铜在肝细胞聚集造成肝细胞坏死,其所含的铜进入血液,然后沉积在脑、肾、角膜等肝外组织而致病。
- 广州金域病理