







| 图片: | |
|---|---|
| 名称: | |
| 描述: | |
- B12-5:第四军医大学附属西京医院
B12-5:第四军医大学附属西京医院
患者女性,61岁,主因“间断头晕10余年,消瘦、下肢浮肿4月”入院。既往患有“高血压病”、“营养性贫血”,并与2012年因“腹部包块”在外院行手术治疗,术后病检回报:腹壁纤维组织细胞及脂肪组织瘤样增生。查体:体温37.5℃,血压120/70mmHg,全身多处可触及大小不一皮下结节,如左侧锁骨上窝、前胸、剑突下、肚脐周围等,右侧乳房近腋窝处可触及4X5cm大的实性包块,无压痛;自身抗体系列、免疫球蛋白系列均阴性。
影像学检查:全身PET-CT提示:1.双侧腋窝、双侧乳腺内前胸、腹壁等部位多个疏松片状软组织病变,双侧下颌骨、双侧锁骨胸骨段、胸骨、脊柱多个椎体及其附件、骨盆、双侧股骨头等多部位弥漫性成骨性病变,均呈葡萄糖代谢异常增高;右心房、升主动脉、腹主动脉周围软组织病变,呈葡萄糖代谢增高;2.左肺上叶舌段结节状高密度病变,葡萄糖轻度异常代谢增高;3.心包膜增厚,心包微量积液。头颅CT示:双侧大脑半球脑外间隙及大脑镰旁散在结节状强化灶。双下肢X线提示双膝关节诸骨骨密度减低,骨小梁稀疏,髓腔内见片絮状高密度影,关节面边缘见骨赘形成,双侧胫腓骨、股骨骨皮质增厚,双膝关节间隙变窄,关节周围见点状骨化影。腹部B超提示双肾大小正常,双肾轻度积水。
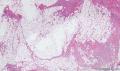
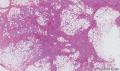
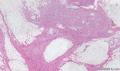
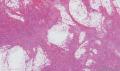
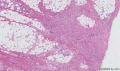
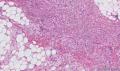
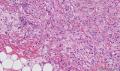
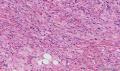
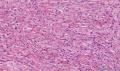
大体取材:右侧腋窝包块:灰黄组织一块,体积:3cmX3cmX2cm,切面可见灰白质硬区,面积3cmX2cm。

名称:图1
描述:b12-5
标签:B12-5 第四军医大学附属西京医院

华夏病理/粉蓝医疗
为基层医院病理科提供全面解决方案,
努力让人人享有便捷准确可靠的病理诊断服务。
×参考诊断
Erdheim-Chester病
-
lantian0508 离线

- 帖子:1250
- 粉蓝豆:42
- 经验:1495
- 注册时间:2007-08-01
- 加关注 | 发消息
出片单位意见: Erdheim-Chester病
-
 snowman103cn: 学习了2014-09-09 23:06
snowman103cn: 学习了2014-09-09 23:06 -
 夏耐午后: 可以详细讲一下吗2014-09-10 09:45
夏耐午后: 可以详细讲一下吗2014-09-10 09:45 -
snowman103cn: 百度一下啊,可清楚了2014-09-10 22:20
华夏病理/粉蓝医疗
为基层医院病理科提供全面解决方案,
努力让人人享有便捷准确可靠的病理诊断服务。
Erdheim-Chester病又称脂质肉芽肿病(Lipogranulomatosis)或脂质肉芽肿瘤样增生病。发病年龄范围7-84岁,中位年龄为53岁数。男性轻度多于女性,主要累及四肢长骨,也可见于扁骨。半数病例伴有内脏损害,包括肾、腹膜后、心脏、心包和肺。临床有局部疼痛及肿胀外,常出现乏力、低热,体重下降,肝脾肿大,还可出现突眼、尿崩症、肾功能衰竭、心肺机能不全,大量出血,神经系统症状及眼睑黄斑。放射影像学表现为双侧骨髓腔斑块状或弥漫性硬化,骨骺变平,1/3病例有溶骨性损害和硬化同时并存。组织学显示大量泡沫细胞、淋巴细胞、浆细胞和杜登细胞浸润,致密纤维组织增生,大块板层骨和网状骨形成。免疫组化Mac387、CD68、CD4、a-AT、a-ACT、S-100阳性表达,不表达Langerhan’s细胞标记[CD1a]。
本病预后不良,大多数病人在3年内死于肾、心血管、肺或中枢神经系统机能不全。
Erdheim-Chester病(ECD)是一种非常罕见的细胞来源不明的非朗格罕组织细胞增多症。
-
夏耐午后: 谢谢赐教!2014-09-10 21:14
-
 watcher035: 学习了2014-09-15 15:37
watcher035: 学习了2014-09-15 15:37 -
 qiaoqiao: 学习了2014-09-25 20:31
qiaoqiao: 学习了2014-09-25 20:31
Erdheim-Chester病(ECD)是一种非常罕见的细胞来源不明的非朗格罕组织细胞增多症。组织细胞增生性疾病包括3个亚型:(1)树突状细胞来源的包括朗格罕组织细胞增多症(LCH )或组织细胞增多症X,Hand-Schller-Christian病和非类脂组织细胞增多。(2) 非朗格罕组织细胞增多症,包括血吞噬淋巴组织细胞增生症,伴有大量淋巴结病的组织细胞增生症和脂肪肉芽肿。(3) 恶性组织细胞增多症。ECD首次由Chester 和Erdheim 于1930年描述,当时命名为脂肪肉芽肿,后以此两人名字冠名该疾病。至今世界范围内有240余例此类疾病被报告,属于罕见病,极易被误诊。
该病好发于中老年人,平均发病年龄在54岁(21~77岁),无性别差异。目前对于这种疾病的发病原因尚不明确,是单克隆增生性疾病还是多克隆反应性疾病仍无定论,有研究表明,ECD与LCH可同时发生,提示两者关系密切或这些组织细胞之间存在相互转化。单核/巨噬细胞和朗罕细胞都是来源于CD34-的骨髓前体细胞。
本病累及范围广泛,最常见的是长骨受累(32%),50%的患者有其他组织浸润,包括皮肤、眶后及眶周组织、下丘脑-垂体轴、心脏、肾脏、神经系统、后腹膜腔、肝、脾、骨骼肌、肾上腺,另外乳腺、甲状腺、睾丸也有个案报告。该疾病的病理镜下特点主要为泡沫样组织细胞增生浸润。20%的患者有肺部受累,表现为不同程度的肺间质纤维化。
ECD的临床表现多样,与病变的部位有关。病变程度可以是轻微的局部受累,也可以是多系统累及。骨痛是最常见的症状,其他症状可有突眼,眼周黄色瘤表现;下丘脑-垂体受累引起的尿崩症是ECD侵犯内分泌系统最常见的临床表现;肾脏受累可出现尿路梗阻甚至肾衰;其他组织受累亦表现为该部位相应功能受损的症状。此外,该疾病还可以伴有发热、虚弱、体重减轻等非特异的表现。
由于该疾病临床表现的多样性和非特异性,极低的发病率和组织学上与其他组织细胞损伤相似导致极易误诊。目前诊断依赖于组织活检,影像学和临床表现的支持。组织活检为确诊依据,典型的组织学表现为:HE染色可见大量泡沫样脂质富集或嗜酸性胞浆的组织细胞;免疫染色以CD68(+),CD1a(-),S-100(+/-)为特征;电镜下Birbeck颗粒缺如。骨影像学可表现为:X线片可见骨髓质缺失,皮质不规则,骨膜增厚;骨闪烁显像技术可见99mTc异常摄取;MRI T1加权相可见散在信号缺失,T2加权相可见高低混合信号。诊断该疾病还需排除一些其他疾病,如朗罕组织细胞增多症(LCH),其特征性免疫标志为CD68(+),CD1a(+), S-100(+),电镜下可见特征性Birbeck颗粒;排除其他代谢性疾病,如黄瘤病等;还需与转移性肿瘤相鉴别。
目前对于ECD的治疗尚存在争议,因为病例数少,对于疗效的观察数据不完善。治疗的方法有:皮质激素、环孢素、干扰素、白血病化疗药物、手术切除、放疗等。
ECD的预后较LCH差,主要与累及的器官有关。呼吸窘迫、广泛的肺组织纤维化、心衰是主要的死亡原因。3年生存率为50%,尚无自行缓解的报告。
学习啦。
-
夏耐午后: 谢谢赐教,受益非浅2014-09-10 21:15
-
watcher035: 学习了2014-09-15 15:37
-
 huocheng_325: 学习了2014-09-23 20:03
huocheng_325: 学习了2014-09-23 20:03 -
qiaoqiao: 谢谢赐教,受益非浅2014-09-25 20:33
-
李建民: 谢谢!学习了!2014-12-31 08:55
引用 8 楼 200406 在 2014-09-10 12:51:15 的发言:
Erdheim-Chester病(ECD)是一种非常罕见的细胞来源不明的非朗格罕组织细胞增多症。组织细胞增生性疾病包括3个亚型:(1)树突状细胞来源的包括朗格罕组织细胞增多症(LCH )或组织细胞增多症X,Hand-Schller-Christian病和非类脂组织细胞增多。(2) 非朗格罕组织细胞增多症,包括血吞噬淋巴组织细胞增生症,伴有大量淋巴结病的组织细胞增生症和脂肪肉芽肿。(3) 恶性组织细胞增多症。ECD首次由Chester 和Erdheim 于1930年描述,当时命名为脂肪肉芽肿,后以此两人名字冠名该疾病。至今世界范围内有240余例此类疾病被报告,属于罕见病,极易被误诊。
该病好发于中老年人,平均发病年龄在54岁(21~77岁),无性别差异。目前对于这种疾病的发病原因尚不明确,是单克隆增生性疾病还是多克隆反应性疾病仍无定论,有研究表明,ECD与LCH可同时发生,提示两者关系密切或这些组织细胞之间存在相互转化。单核/巨噬细胞和朗罕细胞都是来源于CD34-的骨髓前体细胞。
本病累及范围广泛,最常见的是长骨受累(32%),50%的患者有其他组织浸润,包括皮肤、眶后及眶周组织、下丘脑-垂体轴、心脏、肾脏、神经系统、后腹膜腔、肝、脾、骨骼肌、肾上腺,另外乳腺、甲状腺、睾丸也有个案报告。该疾病的病理镜下特点主要为泡沫样组织细胞增生浸润。20%的患者有肺部受累,表现为不同程度的肺间质纤维化。
ECD的临床表现多样,与病变的部位有关。病变程度可以是轻微的局部受累,也可以是多系统累及。骨痛是最常见的症状,其他症状可有突眼,眼周黄色瘤表现;下丘脑-垂体受累引起的尿崩症是ECD侵犯内分泌系统最常见的临床表现;肾脏受累可出现尿路梗阻甚至肾衰;其他组织受累亦表现为该部位相应功能受损的症状。此外,该疾病还可以伴有发热、虚弱、体重减轻等非特异的表现。
由于该疾病临床表现的多样性和非特异性,极低的发病率和组织学上与其他组织细胞损伤相似导致极易误诊。目前诊断依赖于组织活检,影像学和临床表现的支持。组织活检为确诊依据,典型的组织学表现为:HE染色可见大量泡沫样脂质富集或嗜酸性胞浆的组织细胞;免疫染色以CD68(+),CD1a(-),S-100(+/-)为特征;电镜下Birbeck颗粒缺如。骨影像学可表现为:X线片可见骨髓质缺失,皮质不规则,骨膜增厚;骨闪烁显像技术可见99mTc异常摄取;MRI T1加权相可见散在信号缺失,T2加权相可见高低混合信号。诊断该疾病还需排除一些其他疾病,如朗罕组织细胞增多症(LCH),其特征性免疫标志为CD68(+),CD1a(+), S-100(+),电镜下可见特征性Birbeck颗粒;排除其他代谢性疾病,如黄瘤病等;还需与转移性肿瘤相鉴别。
目前对于ECD的治疗尚存在争议,因为病例数少,对于疗效的观察数据不完善。治疗的方法有:皮质激素、环孢素、干扰素、白血病化疗药物、手术切除、放疗等。
ECD的预后较LCH差,主要与累及的器官有关。呼吸窘迫、广泛的肺组织纤维化、心衰是主要的死亡原因。3年生存率为50%,尚无自行缓解的报告。
学习啦。
- 人生就像一场旅行,不必在乎目的地,在乎的是沿途的风景以及看风景的心情,让心灵去旅行。
-
pathology88 离线
- 帖子:56
- 粉蓝豆:0
- 经验:149
- 注册时间:2012-03-24
- 加关注 | 发消息