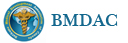





| 图片: | |
|---|---|
| 名称: | |
| 描述: | |
- Case1-淋巴结-欢迎竞猜,一定要来噢!
 图1
图1 图2
图2 图3
图3 图4
图4 图5
图5 图6
图6 图7
图7 图8
图8 图9
图9
| 姓 名: | ××× | 性别: | 男 | 年龄: | 4+月 |
| 标本名称: | 左腋下淋巴结 | ||||
| 简要病史: | 面色苍黄、皮肤瘀点2月,伴低热。入院前拟“嗜血细胞综合征”用激素治疗并抗炎。住院期间PE:患儿躯干、四肢皮肤见针尖样出血点,间有红丘疹,肝脾肋下4厘米,渐增大,左腋下扪及5cm*4cm*4cm肿物。Hb:46-107,WBC 7.0,PLT19-71。出生时有卡介苗接种史。 | ||||
| 肉眼检查: | 融合淋巴结组织一块,3.8cm*2cm*2cm,部分已破,内含淡黄色坏死物,切面为两枚淋巴结,其中一枚完整,切面淡黄。 | ||||
标签:
-
本帖最后由 于 2007-12-25 19:42:00 编辑
×参考诊断
郎格罕细胞组织细胞增生症经过治疗,卡介苗接种后的淋巴结反应性改变
 图1
图1 图2
图2
朋友们好!
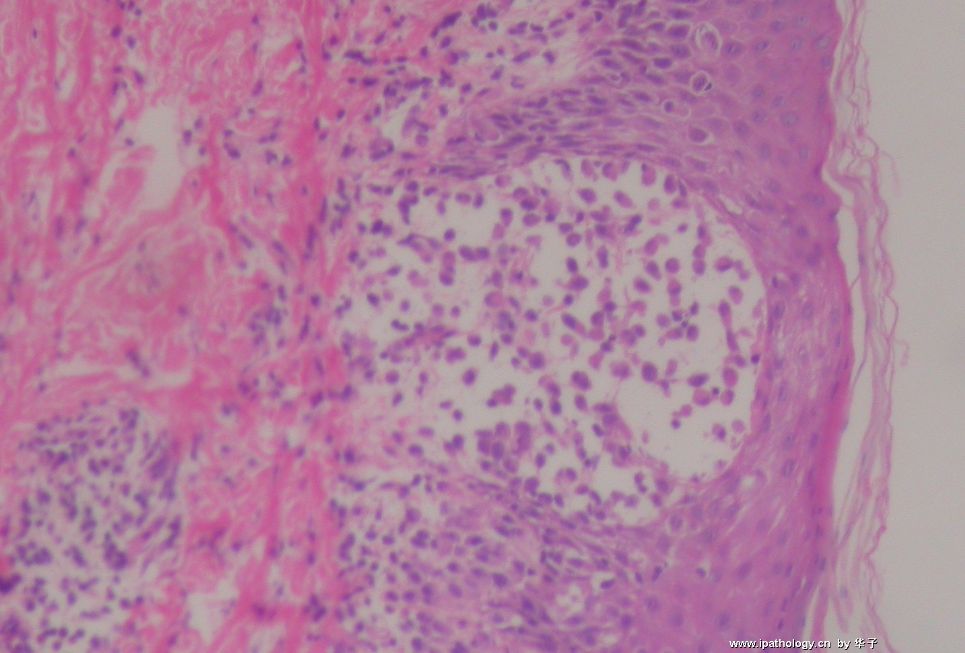
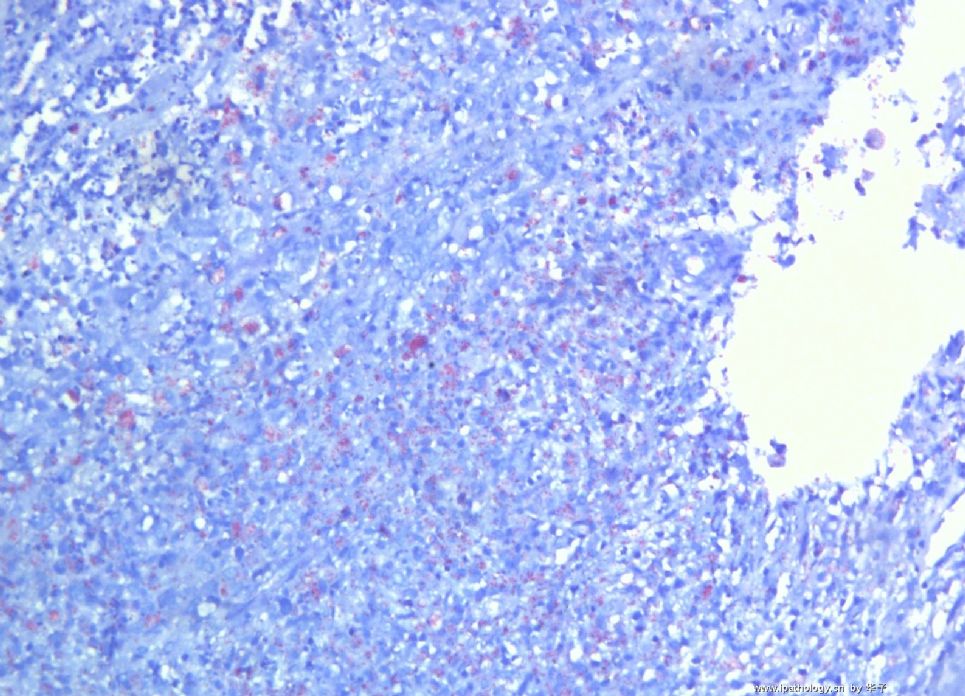
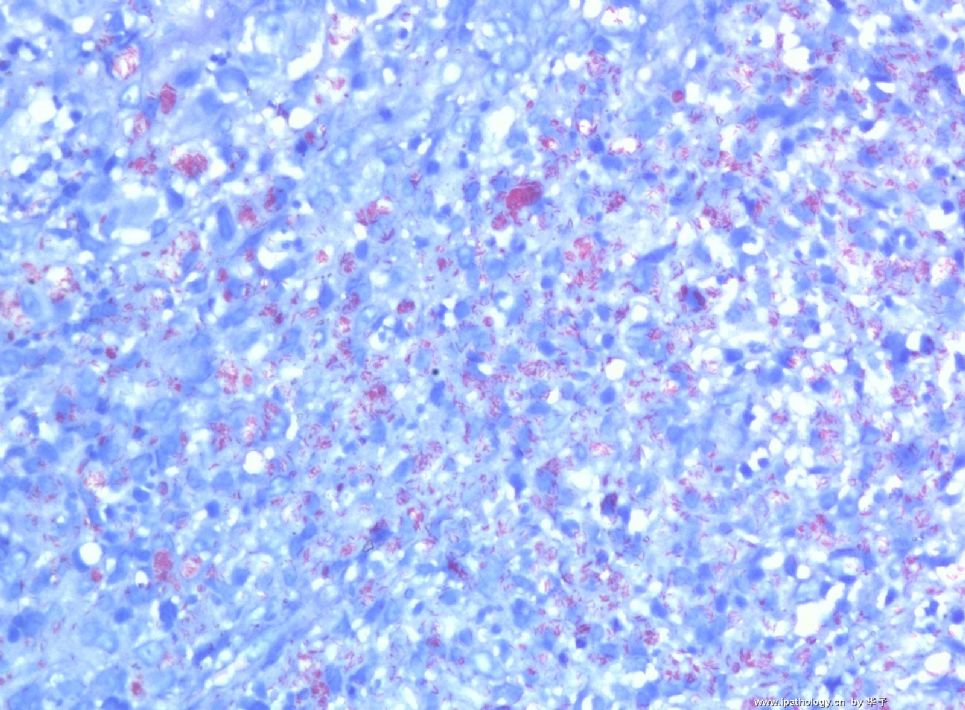
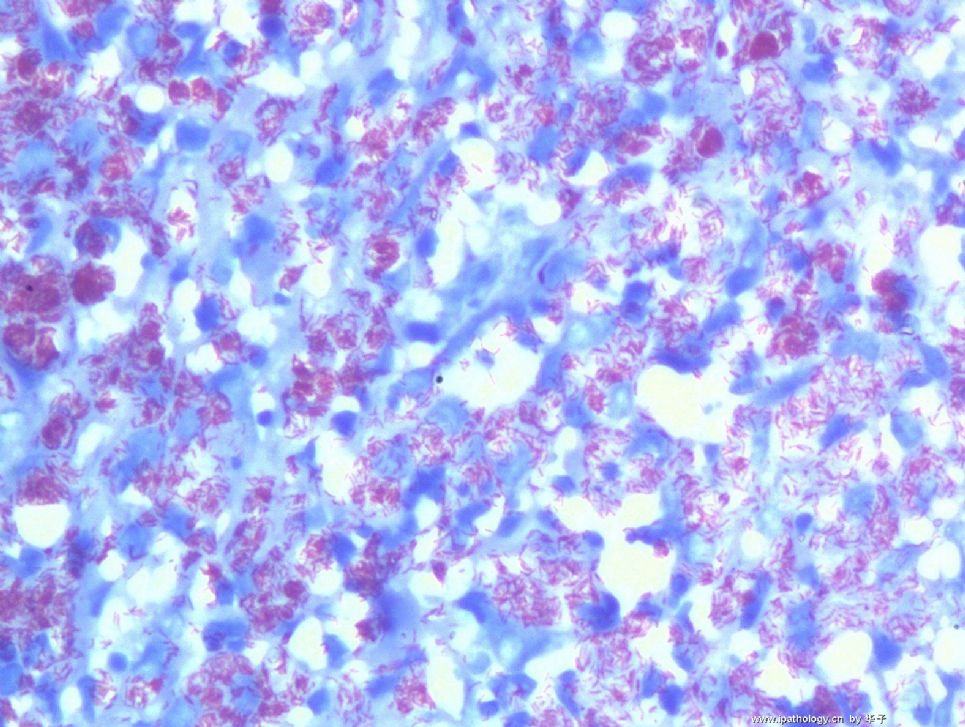
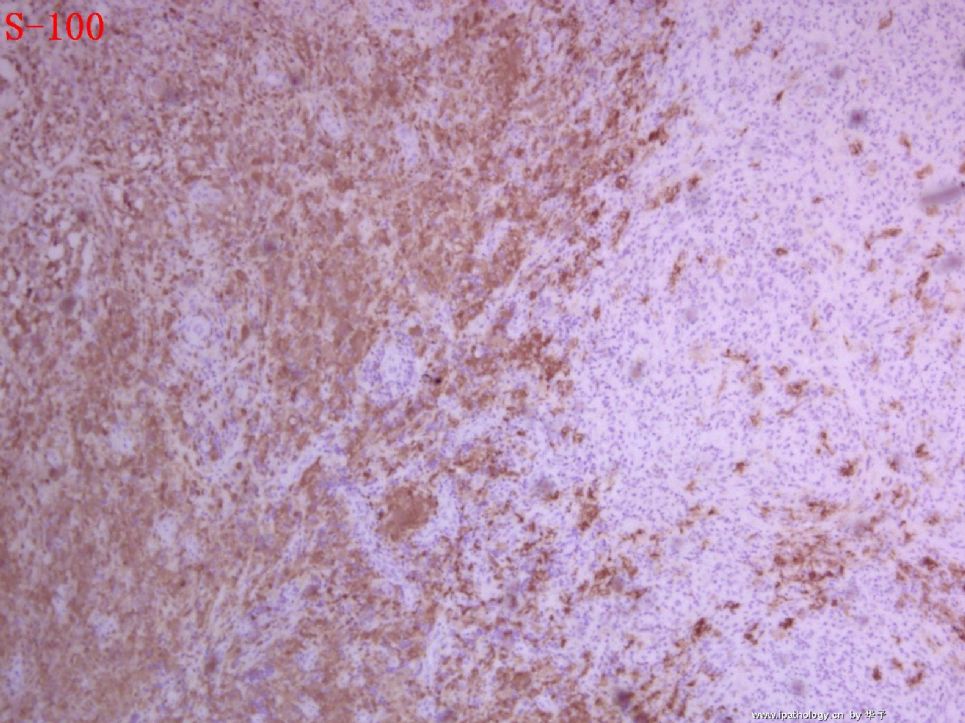
为什么首先选择这个病例是因为我觉得它很有趣,一方面能反映临床病理特点和联系,另一方面能看到因为临床经过导致病变的不典型性。从临床病史来看,本例原发病是郎格罕细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)(Letterer Siwe临床亚型),皮肤、淋巴结均受累,可能累及肝、脾、肺多个器官,皮肤活检亦证实(见插图)。由于长期激素治疗,一方面对LCH起治疗作用,淋巴结病变中的嗜酸性粒细胞少见;另一方面机体免疫机制受到一定影响,导致卡介苗接种后淋巴结强反应,因巨噬细胞功能抑制,虽能吞噬细菌,但不能杀灭细菌,所以胞浆中堆积大量抗酸杆菌;组织虽有坏死,但坏死不彻底;组织细胞虽大量增生,但缺乏类上皮细胞和郎罕巨细胞反应,而呈现泡沫细胞样,见不到典型的结核病样改变。
本例可提供几个复习之处:
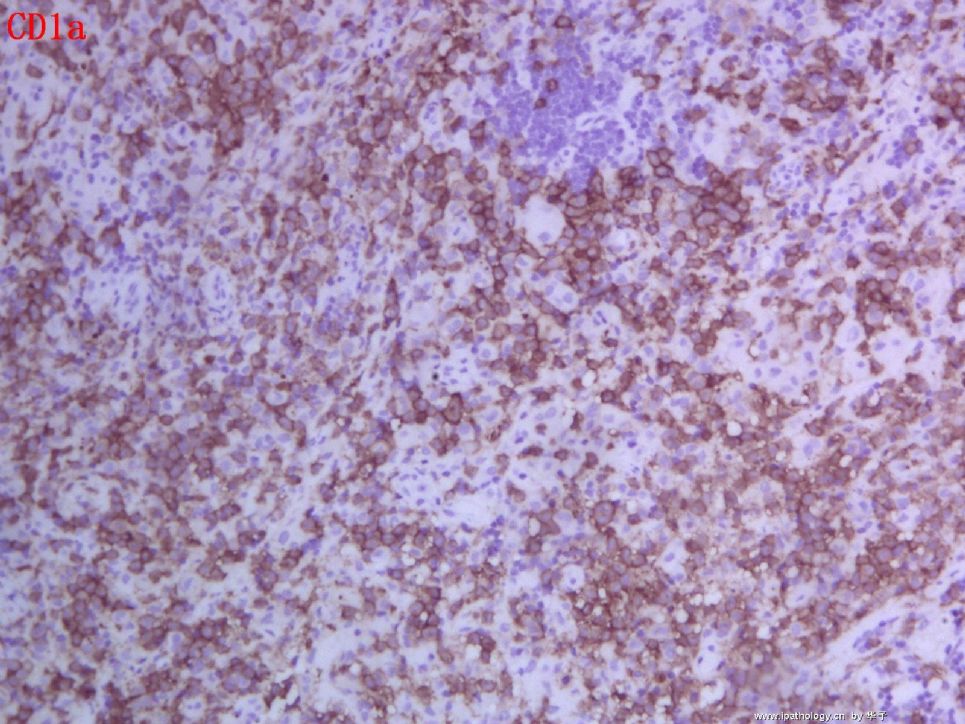
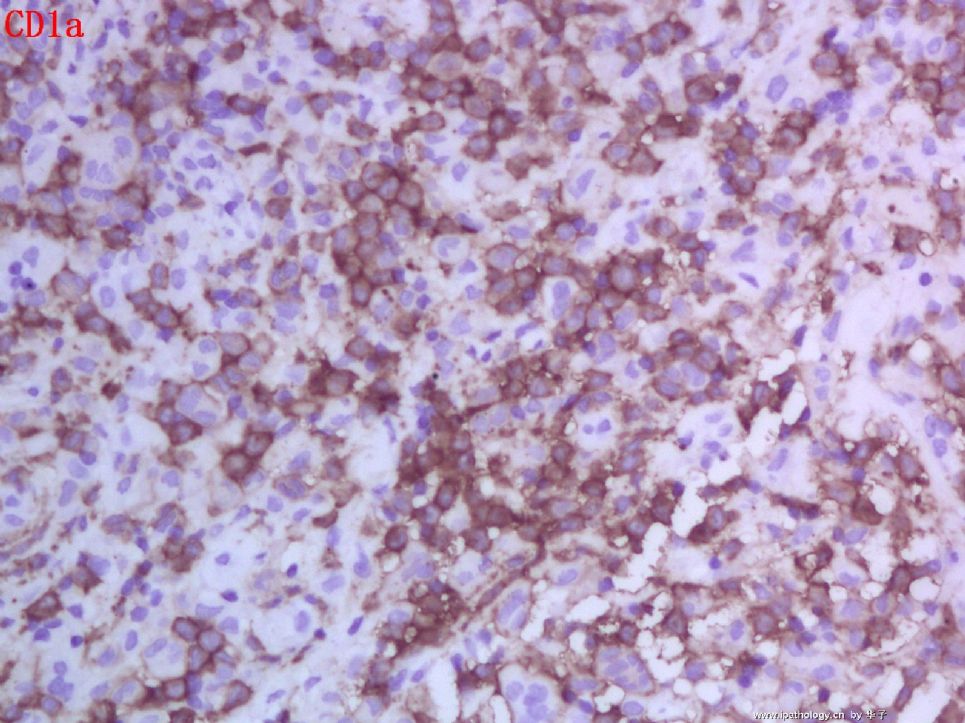
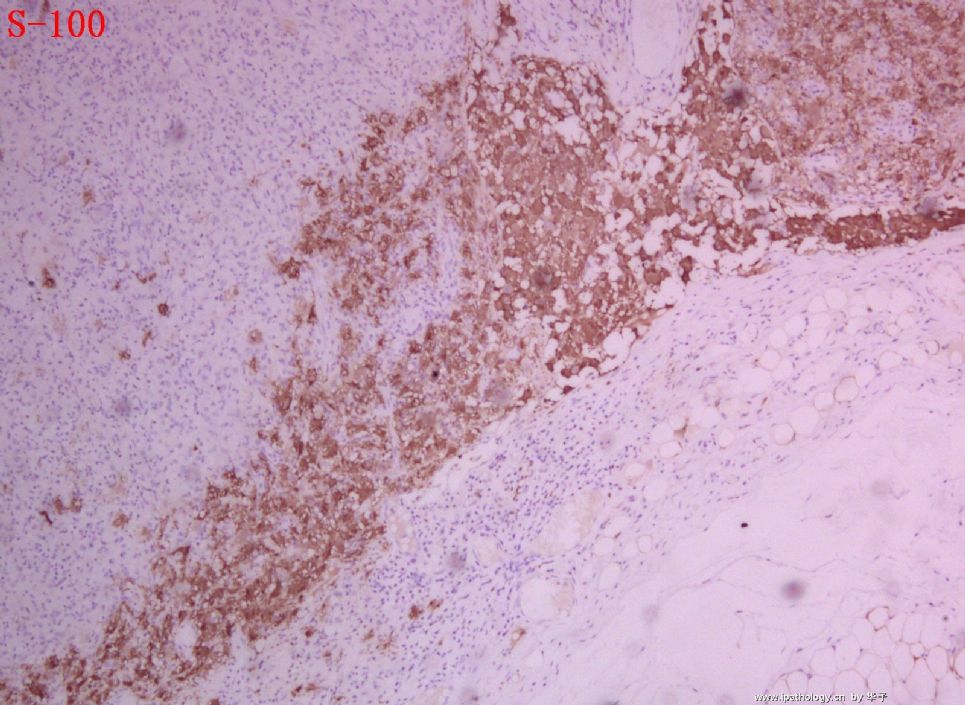
1. 组织细胞(histiocytic cell or phagocyte)及树突状细胞(dendritic cell):WHO将这两种细胞发生的肿瘤归在一起,因为它们分别在抗原处理(antigen-processing)和承递(antigen-presenting)上起作用。两者共同起源于一种骨髓干细胞,但分化方向不同而具有不同的特点和免疫表型,巨噬细胞表达CD68,lysozyme,α-1-antitrypsin,郎格罕细胞表达CD1α,S-100,郎格罕细胞最早发现于皮肤,它可经外周血进入淋巴结,停留于副皮质区成为指状树突细胞。图HE1-4显示组织细胞特点,胞浆丰富,本例因吞噬大量抗酸杆菌而呈浅蓝云雾状,核圆居中,较小,无核沟。HE5-8显示郎格罕细胞特点,胞浆较丰富,红染,核偏位,单核、双核或多核,可见核沟或折叠,略呈肾形或咖啡豆样。
2. LCH的临床分型及特点:LCH定义为表达CD1α和S-100的郎格罕细胞的特发性、肿瘤性增生,特发因病因不明,替代以前的组织细胞增生症X(Histiocytosis X)是因为明确了细胞起源,包括三个临床亚型—勒雪氏病(Letterer-Siwe disease)、韩-苏-克病(Hand-Schuller-Christian disease)和孤立性嗜酸性肉芽肿(Solitary eosinophilic granuloma)。勒雪病常为婴儿急性多灶性、多系统累及,表现发热、皮损、肝脾大、淋巴结肿大、全血减少。我们的部分本型病例平均发病年龄为7个月(N=12,
3. 鉴别诊断噬血细胞综合征(Hemophagocytic syndrome,HPS):又称噬血细胞淋巴组织细胞增生症(hemophagocytic lymphohistiocytosis),是一类少见、死亡率高的疾病。临床表现持续发热、肝脾肿大、肝功能异常、全血减少、凝血障碍、高甘油三酯血症、高细胞因子血症,部分病例伴皮疹,常为一过性伴高热,骨髓、肝、淋巴结等器官内有大量吞噬血细胞的单核巨噬细胞增生。组织学上骨髓或淋巴结中可见大量噬血细胞,胞体大,核偏位,胞浆中吞噬红细胞、血小板、淋巴细胞、粒细胞等。
4. 卡介苗接种反应:卡介苗为牛型减毒活疫菌,经皮内法接种到人体后三个月左右产生免疫力,新生儿6-24小时,无病者均可行卡介苗接种。接种卡介苗一般不引起严重反应,仅个别情况出现局部溃疡和淋巴结肿大、化脓,罕见卡介苗骨髓炎、卡介苗全身播散症等严重异常反应。卡介苗接种后通过淋巴管达全身,接种处附近淋巴结有一定程度的反应,可轻度肿胀,<