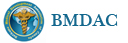。图1")
。图2")
。图3")
。图4")
。图5")
。图6")
。图7")
。图8")
。图9")
。图10")
。图11")











| 图片: | |
|---|---|
| 名称: | |
| 描述: | |
- 一岁男孩胫腓骨内病变,世所罕见(1991至今4例报道,最近到外地学习,又发现了一例,也是小朋友腿上)。
。图1") 图1
图1。图2") 图2
图2。图3") 图3
图3。图4") 图4
图4。图5") 图5
图5。图6") 图6
图6。图7") 图7
图7。图8") 图8
图8。图9") 图9
图9。图10") 图10
图10。图11") 图11
图11
| 姓 名: | ××× | 性别: | 男孩 | 年龄: | 1岁 |
| 标本名称: | 胫骨活检 | ||||
| 简要病史: | |||||
| 肉眼检查: | |||||
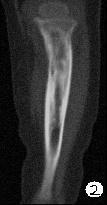
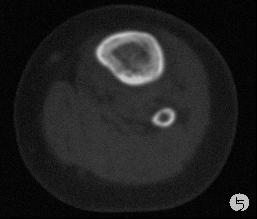
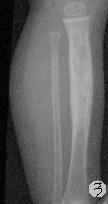
患儿,男,15个月,因被家人发现双下肢不等长并左小腿肿痛4月余,门诊拟诊“左胫骨中上段慢性骨髓炎”收入院。入院4月前,患儿开始学习走路,家人发现其站立不稳,双腿高低不平以及站立时患儿哭啼不适,逐到多家医院就诊,曾被疑为“脑瘫”的可能。外院X光片及CT片提示左胫、腓骨中上段骨病变,以胫骨病变明显,骨皮质不均匀增厚,骨干增粗,局部可见骨质增生及破坏性改变,拟诊“慢性骨髓炎”。入院时,患儿体重10.5Kg,发育正常,营养中等,神志清,精神好,查体合作。专科情况:双下肢不等长,左小腿较右侧长约2cm,局部肌肉无明显改变,中上段压痛,左踝、膝关节活动正常,趾动、血运及感觉正常。入院后在全麻下行左胫骨病灶活检手术。
标签:
-
本帖最后由 于 2010-09-12 22:13:00 编辑
×参考诊断
巨细胞性血管母细胞瘤
Background
This rare soft tissue tumor and presents as a congenital tumor or in early infancy. Less than 5 cases have been described. These tumors have been described in the palate, scalp, and hand. All tumors were ulcerated and may infiltrate the surrounding soft tissue and bone.
INCIDENCE Very rare AGE RANGE-MEDIAN Congenital or early infancy
HISTOLOGICAL TYPES CHARACTERIZATION General Common striking feature are nodular, linear, and plexiform aggregates of oval-to-spindle cells with pink flocculent cytoplasm and round, reniform, or spindle nuclei with dispersed chromatin, small nucleoli, and occasional mitoses
Within these aggregates were large mononucleate cells and multinucleate giant cells with one to several nuclei, often with prominent nucleoli, and abundant granular eosinophilic cytoplasm
Round conglomerates of oval-to-spindle cells and giant cells mimicked non-necrotizing granulomas
Some aggregates had discernible lumens, either empty or containing erythrocytes, lined by plump, cuboidal, or flat endothelial cells
Concentric arrangement of oval-to-spindle cells around small-caliber vascular structures resulted in an onion skin appearance
Envelopment of preexisting blood vessels rarely was observed
Occasional vessels showed eccentric periendothelial proliferation protruding into the lumen
Perineural and intraneural involvement by small vessels was common
In some regions, stroma between the tumoral aggregates was loose, and moderate lymphoplasmacytic infiltrate and scattered mast cells were present
Periphery of the lesions, increase in vessels of capillary or slightly larger size, with plump endothelial cells and generally one or two layers of periendothelial cells
Vessels were separated by a loose stroma containing delicate spindle cellsVARIANTS
SPECIAL STAINS/IMMUNOPEROXIDASE/OTHER CHARACTERIZATION Special stains Immunoperoxidase Factor VIII and CD31 was positive in the endothelium of lesional and nonlesional vessels
Highlighted poorly discernible, small vascular channels within the densely cellular areas
Oval-to-spindle cells in the aggregates and periendothelial cells in the hemangioma-like areas showed strong positive staining for smooth muscle actin and vimentin
Strong positive staining for KP-1 antibody in the multinucleate giant cells and large mononucleate cells
Weakly positive staining for peanut lectin agglutinin in some of the multinucleate giant cells
Negative for:
Desmin, S-100, LCA, EMA, and Cam 5.2
DIFFERENTIAL DIAGNOSIS KEY DIFFERENTIATING FEATURES Giant cell fibroblastoma Express CD34 and show rearrangements of chromosomes 17 and 22 Epithelioid hemangioendothelioma with osteoclast-like giant cells Am J Clin Pathol 1990;93:813?.
Arch Pathol Lab Med 1993;117:315?.
Young adults
Epithelioid tumor cells in these cases, even in solid areas, recapitulated endothelial cells with intracytoplasmic lumina and Factor VIII stainingLack the concentric orientation of cells around small vessels
Plexiform fibrohistiocytic tumor Children and young adults and is characterized by the plexiform and nodular growth of rounded histiocytoid cells with interspersed osteoclast-like giant cell
Onion-skin layering of tumor cells around vessels and the hemangioma-like areas are absent
Myopericytoma Am J Surg Pathol 1998;22:513?5.
Cells with pericytic/myofibroblastic differentiation in a concentric arrangement around vascular lumens
Hemangiopericytoma-like areasGiant cells were not described in cases of myopericytoma
Found mainly in adults, youngest patient was 10 years old
Infantile myofibromatosis, infantile fibromatosis, or infantile fibrosarcoma Oncology Reports 1999;6:1101?.
Am J Surg Pathol 1994;18:14?4.
Characteristic chromosomal aberrations have been described
PROGNOSIS AND TREATMENT CHARACTERIZATION Prognostic Factors Natural history of giant cell angioblastoma is not fully known
Features typically associated with malignancy, such as a brisk mitotic rate, anaplasia, and necrosis are absent in giant cell angioblastoma
The clinical features, histological findings, and follow-up data suggest that giant cell angioblastoma is a benign tumor that would not be expected to metastasize but that could create significant local problems because of its infiltrative nature.
Treatment Am J Surg Pathol 1991;15:175?3.
This tumor extensively infiltrated the skin, soft tissue, and bone of the entire forearm-managed with amputation when the patient was 3 months old
Am J Surg Pathol 2001;25:185-196
With interferon- therapy one had no progression of residual microscopic disease
Second patient experienced considerable regression of the tumor with interferon- administered before subtotal excision-currently has stable minimal residual disease
Am J Surg Pathol 1991;15:175?3.
Am J Surg Pathol 2001;25:185-196

- 广州金域病理
-
本帖最后由 于 2010-03-20 10:31:00 编辑
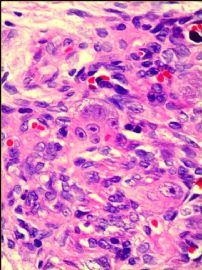 图1
图1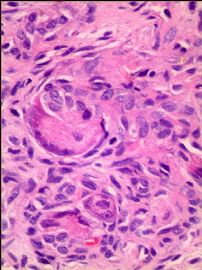 图2
图2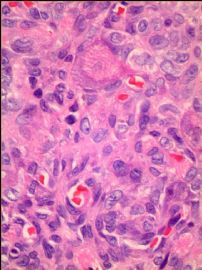 图3
图3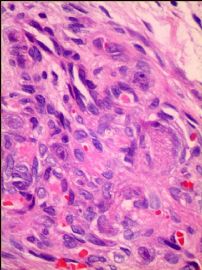 图4
图4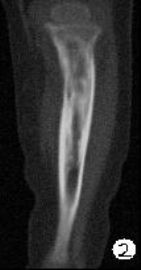 图5
图5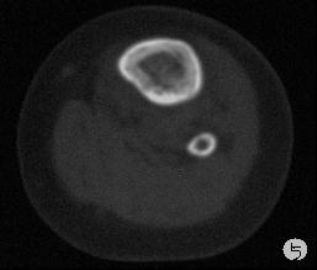 图6
图6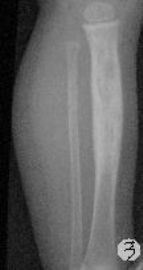 图7
图7
感谢各位专家同行参与的讨论,本病例起病初始,曾在各地就诊,辗转于哈尔滨、北京、深圳的各大医院,考虑骨髓炎等,但均未行手术活检,后到我院骨科住院,行手术取活检,冰冻初诊为簇状血管瘤,冰冻切片组织细胞不明显。常规诊断为“巨细胞血管母细胞瘤”,我们建议用干扰素抗血管治疗,因病例罕见,患者家属要求到各地会诊。国内朱雄增教授会诊确认本病,范钦和教授看过传阅的图片,同意此诊断,陈国璋教授提议:伴有巨细胞血管母细胞瘤形态特征的富于细胞性毛细血管瘤”;但国外几个专家的意见如下:1。Professor Christopher D.M. Fletcher (WHO骨和软组织肿瘤主编,肿瘤组织病理诊断主编) views: Certainly a very unusual and perhaps unique case. There are no more than 5 or 6 convincing examples of giant cell angioblastoma in the English-speaking literature and the "entity" is quite controversial - but it seems that your case could fit
.2。ALLEN: Yes, I agree this is a Giant cell angioblastoma by those pictures. However, I would look at it again to make sure if it is the case, if you could mail me the sections. Giant cell angioblastoma should be a soft tissue tumor and I have never diagnosed or seen a case. Only about 4 in the world literature. It does not appear to be Kaposiform. I don’t know if Chris Fletcher has seen one before. Gonzales-Crussi is the man who would really know. Very interesting. Will be fascinated to hear what Dr Vargas thinks See you in Xian
Phil Allen
3。 this letter is frank gonzalez-crussi to me:
2010/3/2 frank gonzalez-crussi
Dear Dr. Rongjun Mao:
Thank you very much for your letter concerning the boy with a bone lesion that suggested to you the diagnosis of giant-cell angioblastoma. The images that you submitted do resemble, indeed, the lesion that I described originally under that name in the Amer. J. Surg. Pathol. many years ago. I certainly would have made that diagnosis. The original case that I saw was in a child of an age similar to your case, but it involved the arm, and the involvement was so extensive that, as you know from our report, the surgeons decided that an amputation was all they would do. This tumor is very rare. Many pathologists have never seen a case and do not believe it exists.
However, I must tell you that I am now retired, and have been out of the practice of pathology for eight years. The pathologists in the United States that have most experience with this tumor are the group in the Boston Children's Hospital. They have published at least two other cases and have performed extensive studies on the histopathology. They have the most recent information, and probably have important follow-up information about the clinical history of their patients (I remember one of their published cases had caused damage to the palate bones). Dr. Sara Vargas is a pathologist who published the pathology of these cases. You may want to refer your letter to her, and ask her if she has any more information on the clinical evolution of those patients. If you do, please tell her that I referred you to her.
Dr. Sara Vargas e-mail address is:
Sincerely:
F. Gonzalez-Crussi, M.D.
4。 this letter is frank gonzalez-crussi to Chou pauling, :
Dear Pauline:
From the attached letter of Dr.Rongjun Mao (see also the interesting images that he submits) you will see that some people are still concerned about the identification of tumors that we originally described under the name of "giant-cell angioblastoma.' This Chinese doctor saw a case that was identical to the one we saw: the age of the patient was similar, and the site affected was an extremity (in his case a leg, in our case an arm), and the histopathology is identical, with the giant cells around capillary-like formations, and also large gaping cavities. Since I am retired and do not know of anybody with much experience in these lesions, I referred him to Sara Vargas at the Boston Children's Hospital. All the best,
Dear Pauline:
From the attached letter of Dr.Rongjun Mao (see also the interesting images that he submits) you will see that some people are still concerned about the identification of tumors that we originally described under the name of "giant-cell angioblastoma.' This Chinese doctor saw a case that was identical to the one we saw: the age of the patient was similar, and the site affected was an extremity (in his case a leg, in our case an arm), and the histopathology is identical, with the giant cells around capillary-like formations, and also large gaping cavities. Since I am retired and do not know of anybody with much experience in these lesions, I referred him to Sara Vargas at the Boston Children's Hospital. All the best,
5
Thanks Dr. Ma:
I have to collect all the previous cases from Dr. Crussi's file and also some new cases from my hospital I would like to study them more but as you know there's not much tissue left by the time, immunohistochemical studies are performed.
Right now, I only know that the tumor responds to angiogenic therapy. All the cases (with follow-up) has shown good prognosis. I will try to gather the data again and even if you just provide clinical data and follow-up later, it will help others to understand the tumor better. Thanks again for sharing with us and I will keep you informed.
If you have some unstained slides, or touch preps, we could still try to perform some tests. Keep in touch and I will give you the follow-up.
Sincerely,
Pauline
巨细胞血管母细胞瘤(Giant Cell Angioblastoma,GCAB)是一种极其罕见的具有局部侵袭性的软组织肿瘤, 由F. Gonzalez-Crussi等于1991年首先报道,之后于2001年Sara O. Vargas等又报道了一组3例的病例。虽然报道的病例不多,但因这种病变具有独特的形态学表现,因此GCAB已被接受为一个独立的病理实体 。尽管如此,基于病例报道的有限性,GCAB是否真正代表了一种血管性肿瘤实体尚存质疑,需要获得更多的病例资料才能对其诊断标准和生物学潜能进行界定,因此在2002版WHO肿瘤新分类中尚未接纳这一实体作为新的肿瘤分类类型。该肿瘤具有如下特征性的形态学表现:肿瘤呈结节状、粱索状或丛状,由卵圆形至梭形的瘤细胞混杂数量不等的单核组织细胞样巨细胞及多核巨细胞呈同心圆样排列在具有血管内皮细胞衬里的小血管腔隙周围形成洋葱皮样外观。本瘤罕见,遇到时可能将其误诊为其它各种类型的肿瘤。
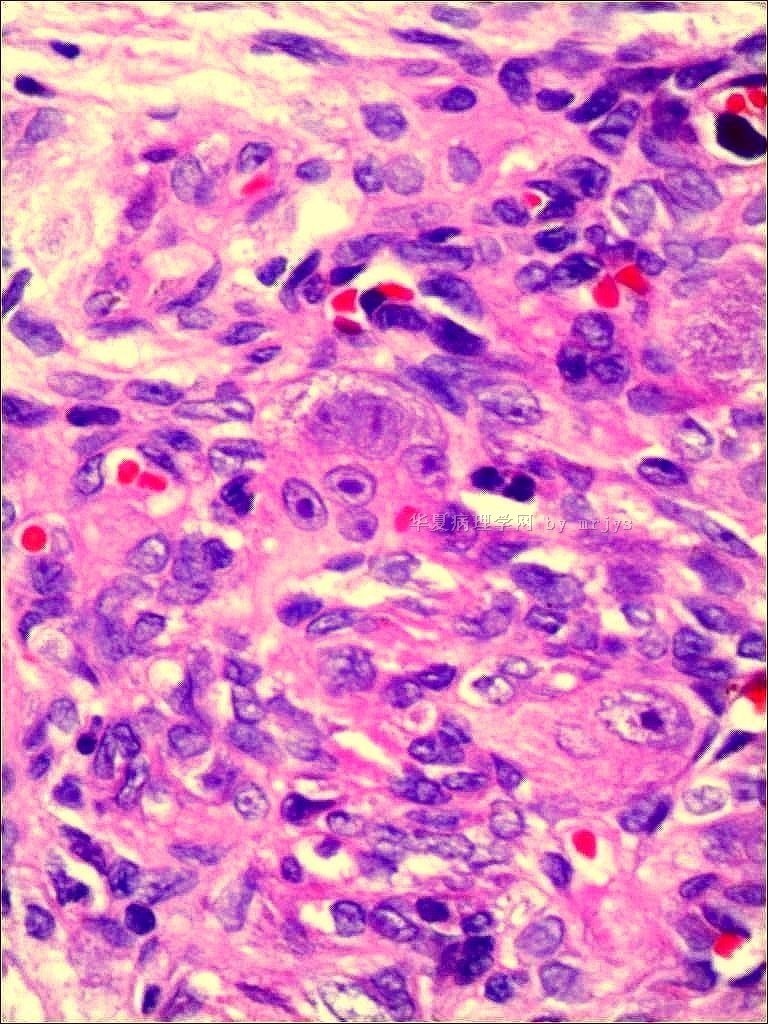
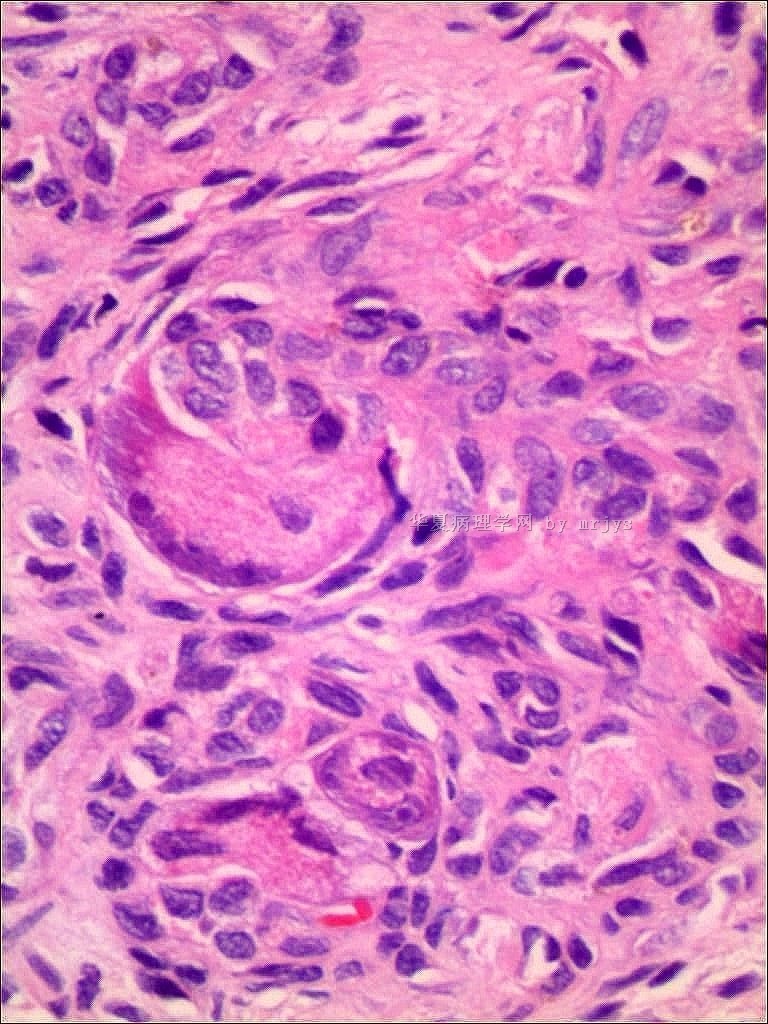
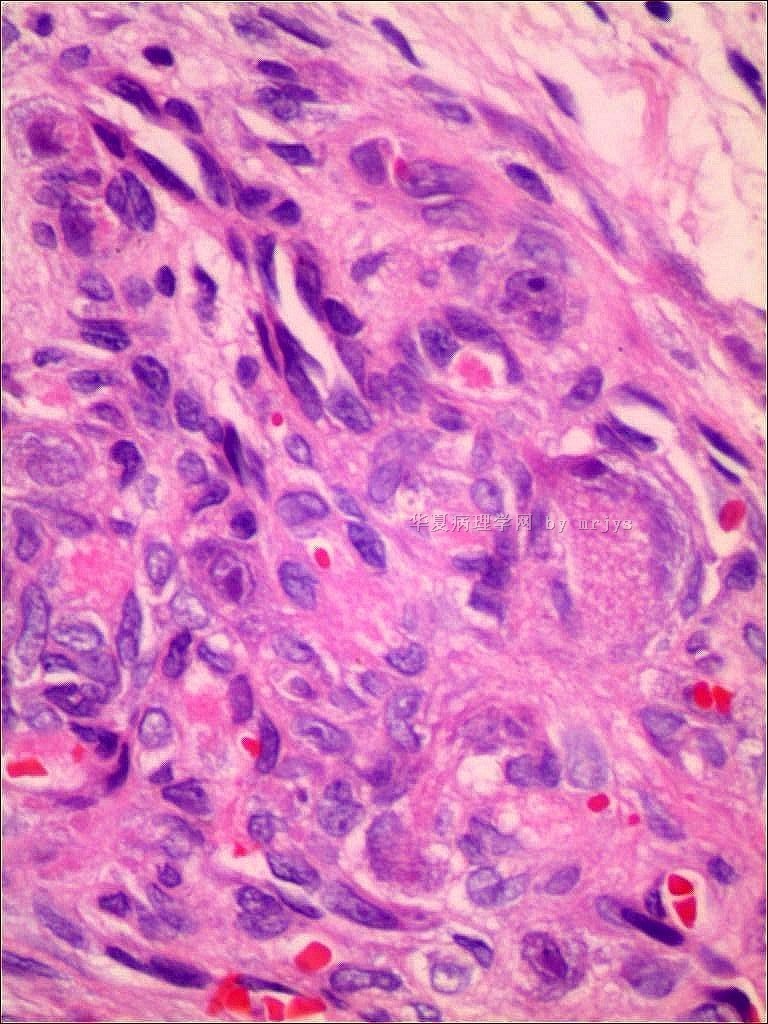
我们的病例镜下:病变组织位于边缘被覆少量骨母细胞的纤细的骨小梁之间并取代了正常增生活跃的骨髓组织,肿瘤组织由结节状、粱索状或丛状的富于卵圆形至梭形的瘤细胞构成,这些区域可见开放或未开放的微血管腔隙,部分腔隙内含有一到数个红细胞, 在卵圆形至梭形瘤细胞之间散在多少不等的单核组织细胞样巨细胞及多核巨细胞,这些区域形态类似于非坏死性肉芽肿或纤维组织细胞瘤样病变,少量结节内瘤细胞围绕在微血管腔隙周围呈同心圆样排列,加之外周胶原纤维的包绕形成洋葱皮样外观;在少数瘤细胞聚集的边缘,毛细血管扩张并充满红细胞或血清蛋白样物质,形态近似海绵状血管瘤;在临近瘤细胞聚集的周围间质中常常可以见到一条或多条厚壁中小血管穿越;部分瘤细胞聚集的区域缺乏单核组织细胞样巨细胞及多核巨细胞,这时形态类似富于细胞性血管瘤、血管周细胞瘤或丛状血管瘤样结构;卵圆形至梭形瘤细胞多数肥胖,胞浆丰富红染,核膜厚,染色质细颗粒状,多数可见一个小核仁;另见少量瘤细胞呈退行性改变,表现为细胞核缩小,染色质浓缩,甚至消失;单核组织细胞样巨细胞及多核巨细胞具有相同的细胞学形态,细胞巨大,可达周围卵圆形至梭形瘤细胞三至十倍横径大小,胞浆丰富,颗粒状,嗜酸性或嗜双色性,部分胞浆嗜碱性,核膜厚,核呈空泡状,具有一个显著的粗大核仁,单核组织细胞样巨细胞核位于中央,多核巨细胞的核多数排列成半花环状或者马蹄状,也有少量巨细胞类似于杜顿巨细胞或破骨细胞样巨细胞,少数巨细胞亦具有退变的倾向及胞浆内空泡形成。另外,部分相互毗邻的卵圆形至梭形瘤细胞胞相互靠拢,细胞核呈环状排列,中央为融合的胞质,类似花环状多核巨细胞,但缺乏粗大的核仁,部分融合的胞浆逐步空泡化形成环状血管样腔隙,其中可有少量的红细胞滞留;所有肿瘤细胞聚集区域未发现核分裂相、坏死及细胞的非典型性;肿瘤间质疏松,可见稀疏形态温和的梭形星形细胞、单核炎症细胞及少量肥大细胞,间质内未发现单核组织细胞样巨细胞及多核巨细胞。另外,尚可见少量骨母细胞细胞增生活跃的骨组织。
快速冰冻切片组织镜下形态与石蜡切片组织相近,瘤组织排列成结节状、粱索状或丛状,但其中单核组织细胞样巨细胞及多核巨细胞不易辨认,术中冰冻切片诊断意见:分叶状良性肿瘤,考虑为细胞性血管瘤或丛状血管瘤,待常规石蜡切片进一步分型。
免疫组化:肿瘤实质内卵圆形至梭形瘤细胞、单核组织细胞样巨细胞、多核巨细胞及间质各成分弥漫性强阳性表达Vimentin;部分卵圆形至梭形瘤细胞包括构成微血管样腔隙及环状血管样腔隙的内皮细胞样细胞强表达CD34及CD31,灶状表达FⅧ;单核组织细胞样巨细胞及多核巨细胞强表达CD68,偶见个别巨细胞胞浆内局灶性表达FⅧ;发育良好的血管及裂隙状小血管外周细胞a-SMA强阳性,而其他区域瘤细胞呈弱阳性表达,内皮细胞样细胞阴性;局灶性瘤细胞聚集区散在少量的小梭形或多边形细胞表达S100;间质内发育良好的中、小血管壁周细胞强阳性表达Calponin和h-Caldesmon,而其他组织不表达;EMA、AE1/AE3、CD1a肿瘤实质及间质均未见表达。
GCAB是一种极其罕见的肿瘤,加之本文提供的病例,目前仅有5例报道,其中男性3例,女性2例;发病年龄从出生到一岁以内的婴幼儿,可累及皮肤、粘膜、皮下软组织及骨组织;病变呈红斑、水泡、结节、浸润性斑块及溃疡性改变,所有病例均具有局部侵袭性的特征。我们提供的这一病例,为男性婴儿,X光片及CT片显示病变位于左侧胫骨、腓骨中上段,以胫骨改变明显,同时具有骨质增生和破坏性改变的特点,影像学医生可能考虑为“慢性骨髓炎”或“纤维结构不良”。另外,由于病变位于左下肢骨内,临近的皮肤、软组织及关节无特殊改变,故病变早期难于被发现,甚至以“无法站立或站立不稳”就诊时曾被疑为“脑瘫”。
鉴别诊断:需要与GCAB 做鉴别诊断的病变包括获得性丛状血管瘤(Acquired Tuffted Hemangioma,ATH)、富于细胞性毛细血管瘤(Cellular Capillary Hemangioma,CCH)、卡波西样血管内皮瘤(Kaposiform Haemangioendothelioma,KH)、巨细胞纤维母细胞瘤(Giant Cell Fibroblastoma,GCF)、伴有破骨样巨细胞的上皮样血管内皮瘤(Epithelioid Hemangioendothelioma with Osteoclastlike Giant Cells,EH with OGC)、巨细胞血管纤维瘤(Giant Cell Angiofibroma,GCAF)、血管瘤样纤维组织细胞瘤(Angiomatoid Fibrous Histiocytoma,AFH)、多核细胞血管组织细胞瘤(Multinucleate Cell Angiohistiocytoma, MCA)、丛状纤维组织细胞瘤(Plexiform Fibrohistiocytic Tumor,PFT)、先天性肌纤维瘤病(Congenital Myofibromatosis,CMF等。GCAB肿瘤实质呈结节状、梁索状及丛状,这些瘤细胞聚集区主要由卵圆形至梭形瘤细胞构成并散在数量不等的单核组织细胞样巨细胞及多核巨细胞,这些瘤细胞聚集区可见数量不等的微血管腔隙,少量结节形态类似洋葱皮样外观。某些区域的形态可能与ATH、CCH及KH相混淆,但后三者一致性缺乏核仁醒目的单核组织细胞样巨细胞及多核巨细胞,易于鉴别。值得注意的是,对于本病例,尽管有人提议采用两个新的术语“伴有巨细胞血管母细胞瘤形态特征的获得性丛状血管瘤(Acquired Tuffted Hemangioma with morphologic features of Giant cell Angioblasttoma,GCABATH” 或“伴有巨细胞血管母细胞瘤形态特征的富于细胞性毛细血管瘤(Celllular Capillary Hemangioma with morphologic features of Giant cell Angioblasttoma, GCABCCH)”,因目前尚无文献报道的先例,我们最终未予采纳,而GCAB尽管病例很少,但其形态学特征与本病例的形态极其贴近,因此我们认为,对于本病例的诊断宜采用术语GCAB更为恰当。GCF由梭形细胞和不规则分布、内衬核大深染多核巨细胞的裂隙样或血窦样假脉管性腔隙构成,间质粘液样或胶原化,这种巨细胞与GCAB中巨细胞具有显著差别,后者细胞核呈圆形或卵圆形,核膜厚且清晰,胞浆空淡,还有一个大核仁,有利两者的区别;EH with OGC发生于青年人,与大血管有关,除了破骨样巨细胞以外,上皮样内皮细胞具有胞浆内腔隙及表达FVIII,不同程度的细胞非典型性及核分裂像、而GCAB,发生于婴幼儿,瘤细胞具有间充质细胞、纤维母细胞、肌纤维母细胞、周细胞及内皮细胞的特征,瘤细胞无核分裂像、非典型性及坏死,加上醒目的巨细胞,两者不难鉴别;GCAF特征性形态表现为肿瘤富含血管,并含有不规则分布的血窦样或裂隙样假血管性腔隙,内衬一层不连续的核深染多核巨细胞,类似GCF,但腔隙周围常有出血,肿瘤的部分区域类似孤立性纤维瘤,卵圆形或胖梭形细胞和异型多核巨细胞表达CD34,而GCAB单核组织细胞样细胞及多核巨细胞表达CD68,不表达CD34并缺乏类似孤立性纤维瘤样区域。AFH肿瘤组织由成片组织细胞样细胞和囊状扩张的出血性假血管腔隙组成,肿瘤的周边常伴有炎症细胞浸润,并常具有致密的包膜,类似残留的淋巴结,而GCAB肿瘤结节内血管腔隙微小,开放或未开放,缺乏囊状扩张的出血性假血管腔隙,以及肿瘤缺乏假包膜,周边缺乏淋巴细胞套样结构;MCA是一种好发于40岁年龄的皮肤丘疹或结节性病变,主要病理改变为真皮内血管增生伴胶原间穿插圆形卵圆形单核组织细胞样巨细胞及散在不规则状或锯齿状多核巨细胞。单核组织细胞不形成结节,多核巨细胞散在致密的胶原性间质中,不像GCAB,后者发病年龄小,瘤细胞形成结节状、粱索状及丛状,间质疏松水肿。PFT由丛状分布的多个小结节或梭形细胞束组成,结节由单核组织细胞样巨细胞、梭形纤维母细胞样细胞和破骨细胞样多核巨细胞混杂而成。单核组织细胞样巨细胞、梭形纤维母细胞样细胞胞具有细胞的非典型性,巨细胞为破骨样巨细胞,巨细胞缺乏醒目的大核仁,肿瘤实质与间质没有截然分界,而GCAB单核组织细胞样巨细胞与多核巨细胞大小一致,核空泡状明显,核仁醒目,各种细胞成分无非典型性,肿瘤实质与间质分界清楚,后者疏松水肿;CM好发于男性婴幼儿,特点为多发性坚实性真皮和皮下结节,组织病理显示肿瘤呈结节状,富于细胞区显示有卵圆形到梭形核的成纤维细胞排列成交织束状或漩涡状,在细胞较少区,间质可呈黏液样和毛细血管增生,缺乏GCAB肿瘤组织中的单核组织细胞样巨细胞及多核巨细胞,以资鉴别。
本病例累及左侧胫骨、腓骨中上段,病变范围大,多发,界限不清,病变骨组织具有增生性和破坏性改变,进一步证实了GCAB是一种罕见的发生于婴幼儿,具有独特形态学特征且局部具有侵袭性的血管源性肿瘤。对于GCAB的治疗,需尽可能把肿瘤组织完整切除并确保其切缘阴性,当广泛累及肢体或多次复发者需考虑截肢手术。由于GCAB是一种血管源性肿瘤,这为抗血管生成治疗提供了理论基础,先前报道的两例患者经过干扰素抗血管治疗后随访发现已取得良好的疗效。因此我们建议,对于肿瘤不能切除或切除不完整的病例,应尝试应用干扰素治疗。我们提供的这例病例因发生于左小腿胫、腓骨中上段骨髓腔内,病变相对广泛,不易手术切除,我们推荐患儿进一步接受干扰素治疗以期取得良好的效果。