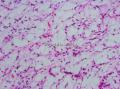
复旦大学儿科医院提供图1")
复旦大学儿科医院提供图2")
复旦大学儿科医院提供图3")
复旦大学儿科医院提供图4")
复旦大学儿科医院提供图5")
复旦大学儿科医院提供图6")
复旦大学儿科医院提供图7")

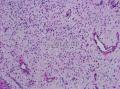






| 图片: | |
|---|---|
| 名称: | |
| 描述: | |
- 颈部肿块-上海市骨与软组织肿瘤读片2013(2-1)复旦大学儿科医院提供
-
本帖最后由 海上明月 于 2013-09-14 18:12:00 编辑
1. J Pediatr Hematol Oncol. 2013 May 9. [Epub ahead of print]
Saito A, Taketani T, Kanai R, Kanagawa T, Kumori K, Yamamoto N, Ishikawa N, Takita J, Yamaguchi S.
Source
Departments of *Pediatrics §Radiology ∥Organ Pathology, Shimane University Faculty of Medicine †Division of Blood Transfusion, Shimane University Hospital ‡Department of Digestive and General Surgery, Shimane University School of Medicine, Shimane ¶Department of Cell Therapy and Transplantation Medicine, Graduate School of Medicine, University of Tokyo, Tokyo, Japan.
Abstract
A girl, aged 19 months, presented with a sacrococcygeal tumor that developed at 5 months after birth and gradually enlarged. Serum tumor marker levels were negative. Ultrasound imaging showed abundant blood flow in the tumor. However, neither computed tomography (CT) nor magnetic resonance imaging (MRI) showed contrast agent incorporation. The surgically resected tumor consisted of immature cells with myxoid stroma and proliferating small blood vessels. Immunostaining showed extensive vimentin expression. However, smooth muscle actin, muscle-specific actin, and S-100 protein expression was negative. Neither the ETV6-NTRK3 fusion gene nor the FUS gene rearrangement was detected. Thus, the patient was diagnosed with a primitive myxoid mesenchymal tumor of infancy. This tumor primarily consisted of a mucosal stroma with a low absorption on CT, a low signal on T1-weighted MRI, and a high signal on T2-weighted MRI. A diagnosis of primitive myxoid mesenchymal tumor of infancy should be considered in cases of soft tissue tumors in infants that show prominent vascularity but little contrast enhancement on MRI or CT.
2. Med Mol Morphol. 2013 Jun;46(2):109-13. doi: 10.1007/s00795-013-0032-1. Epub 2013 Mar 5.
Su TC, Hwang MJ, Li CF, Wang SC, Lee CH, Chen CJ.
Source
Department of Surgical Pathology, Changhua Christian Hospital, 135 Nanxiao Street, Changhua, Taiwan.
Abstract
Primitive myxoid mesenchymal tumor of infancy is an extremely rare and recently recognized soft tissue tumor entity with a tendency for multiple recurrences. Only ten cases have been described in the literature and most cases are reported in Western countries. This tumor ranges in size from 2 to
3. Pathol Int. 2012 Aug;62(8):549-53. doi: 10.1111/j.1440-1827.2012.02836.x. Epub 2012 Jun 21.
Primitive myxoid mesenchymal tumor of infancy: report of two cases and review of the literature.
Source
Department of Pathology, First Affiliated Hospital of Nanjing Medical University, Nanjing, China.
Abstract
Primitive myxoid mesenchymal tumor of infancy is a recently recognized soft tissue tumor with only a few cases reported. Here, we reported another two cases of the lesion, a 5-month-old boy presenting with a soft tissue mass in the neck region that recurred 2 months later and a 3-day-old girl with a congenital superficial dorsal lumbar mass that extended to the spinal canal 1 month later. They shared similar histological patterns, such as unusual diffuse myxoid background, delicate vascular network, small cystic spaces, low to moderate cellularity, and primitive mesenchymal tumor cells. Immunohistochemically, the tumor cells showed positive for vimentin, CD99, CD117 and nestin, negative for myoid, lipoblastic, histiocytic, and neural markers. In conclusion, primitive myxoid mesenchymal tumor of infancy is a distinctive entity with its own clinical pathological features. Expression of CD99, CD117 and nestin may be consistent with the primitive nature of the tumor and may serve as ancillary markers for differential diagnosis from the other infantile tumors.
4. Pediatr Dermatol. 2010 Nov-Dec;27(6):635-7. doi: 10.1111/j.1525-1470.2010.01323.x. Epub 2010 Nov 16.
Primitive myxoid mesenchymal tumor of infancy in a preterm infant.
Lam J, Lara-Corrales I, Cammisuli S, Somers GR, Pope E.
Source
Department of Pediatrics, Department of Dermatology, University of British Columbia, Vancouver, British Columbia, Canada. joseph.mc.lam@gmail.com
Abstract
Primitive myxoid mesenchymal tumor of infancy is a recently recognized entity that has been added to the differential diagnosis of myxoid tumors of the soft tissue. Few cases have been reported of this entity in the literature, but none presenting in a preterm infant. We present the case and clinical course of a preterm boy with a primitive myxoid mesenchymal tumor of infancy that occurred following excision of a congenital juvenile xanthogranuloma.
5. Pediatr Dev Pathol. 2011 Jan-Feb;14(1):75-9. doi: 10.2350/
Mulligan L, O'Meara A, Orr D, Eadie P, Hayes R, McDermott M.
Source
Department of Histopathology, Our Lady's Children's Hospital Crumlin, Dublin, Ireland.
Abstract
We report a case of an 8-month-old child with a primitive myxoid mesenchymal tumor of infancy arising in the thenar eminence. The lesion recurred after conservative excision and was ultimately nonresponsive to chemotherapy, necessitating partial amputation. The patient remains free of disease 5 years after this radical surgery. This is the 1st report of such a tumor since it was initially described by Alaggio and colleagues in 2006. The pathologic differential diagnosis is discussed.
6. Am J Surg Pathol. 2006 Mar;30(3):388-94.
Primitive myxoid mesenchymal tumor of infancy: a clinicopathologic report of 6 cases.
Alaggio R, Ninfo V, Rosolen A, Coffin CM.
Source
Department of Oncology and Surgery, Section of Pathology, University of Padova, Padova, Italy.
Abstract
Soft tissue sarcomas in the first year of life are rare, and the most common sarcomas in infancy are embryonal rhabdomyosarcoma, Ewing sarcoma/primitive neuroectodermal tumor, congenital infantile fibrosarcoma, and primitive sarcomas such as undifferentiated sarcoma. In this study, we report 6 cases of a primitive myxoid mesenchymal tumor of infancy (PMMTI), which previously may have been included under the diagnostic categories of congenital-infantile fibrosarcoma or infantile fibromatosis. PMMTI occurred in 6 infants, 3 of whom had a congenital presentation of a soft tissue mass. All patients were otherwise healthy. The tumors occurred on the trunk, extremities, and head and neck. Grossly, the tumors were nonencapsulated and had a multinodular appearance with focal infiltrative growth, a white fleshy cut surface, and a tumor diameter ranging from 2 to

- 王军臣
-
本帖最后由 海上明月 于 2013-09-14 18:10:32 编辑
1. Am J Surg Pathol. 2006 Mar;30(3):388-94.
Primitive myxoid mesenchymal tumor of infancy: a clinicopathologic report of 6 cases.
Alaggio R, Ninfo V, Rosolen A, Coffin CM.
Source
Department of Oncology and Surgery, Section of Pathology, University of Padova, Padova, Italy.
Abstract
Soft tissue sarcomas in the first year of life are rare, and the most common sarcomas in infancy are embryonal rhabdomyosarcoma, Ewing sarcoma/primitive neuroectodermal tumor, congenital infantile fibrosarcoma, and primitive sarcomas such as undifferentiated sarcoma. In this study, we report 6 cases of a primitive myxoid mesenchymal tumor of infancy (PMMTI), which previously may have been included under the diagnostic categories of congenital-infantile fibrosarcoma or infantile fibromatosis. PMMTI occurred in 6 infants, 3 of whom had a congenital presentation of a soft tissue mass. All patients were otherwise healthy. The tumors occurred on the trunk, extremities, and head and neck. Grossly, the tumors were nonencapsulated and had a multinodular appearance with focal infiltrative growth, a white fleshy cut surface, and a tumor diameter ranging from 2 to
2. Genes Chromosomes Cancer. 1996 Feb;15(2):115-21.
Kaneko Y, Yoshida K, Handa M, Toyoda Y, Nishihira H, Tanaka Y, Sasaki Y, Ishida S, Higashino F, Fujinaga K.
Source
Department of Cancer Chemotherapy, Saitama Cancer Center Hospital, Japan.
Abstract
EIAF is a newly isolated ETS-family gene that is located on 17q21 and codes for the adenovirus EIA enhancer-binding protein. In our chromosome analysis of 18 of the Ewing family of tumors and undifferentiated sarcomas, we found t(17;22)(q12;q12) in an MIC2 antigen-positive undifferentiated sarcoma of infancy. On Southern blot analysis, EWS and EIAF cDNA probes hybridized to the same rearranged band, indicating that an EWS-EIAF fusion gene was formed in the tumor. Further Southern blot analysis using four EIAF cDNA probes of different sizes showed that the breakpoint lies in the region upstream to the ETS domain of the EIAF gene. EIAF may be the fourth ETS-family gene to be identified forming a fusion gene with EWS. We assume that the RNA binding domain of EWS may have been replaced by the DNA binding domain of EIAF in the EWS-EIAF fusion protein as in other fusion proteins previously characterized in Ewing sarcoma and other types of sarcomas.
3. J Am Coll Cardiol. 1985 Dec;6(6):1362-4.
Intracardiac undifferentiated sarcoma in infancy.
Ludomirsky A, Vargo TA, Murphy DJ, Gresik MV, Ott DA, Mullins CE.
Abstract
A rare case of an intracardiac undifferentiated sarcoma in a 3 month old infant is described together with the clinical, angiographic, echocardiographic, surgical and histopathologic findings. The tumor was successfully removed surgically, and monthly echocardiographic follow-up is being performed.
- 王军臣
-
snowman103cn 离线

- 帖子:518
- 粉蓝豆:9
- 经验:538
- 注册时间:2010-11-07
- 加关注 | 发消息
-
xiaofeng1008 离线
- 帖子:783
- 粉蓝豆:33
- 经验:824
- 注册时间:2013-07-24
- 加关注 | 发消息