







 好病例,很有迷惑性!
好病例,很有迷惑性! | 图片: | |
|---|---|
| 名称: | |
| 描述: | |
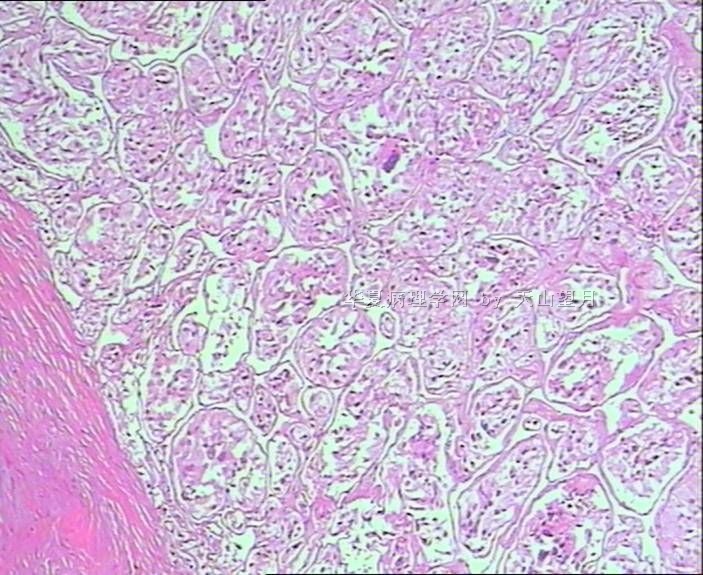
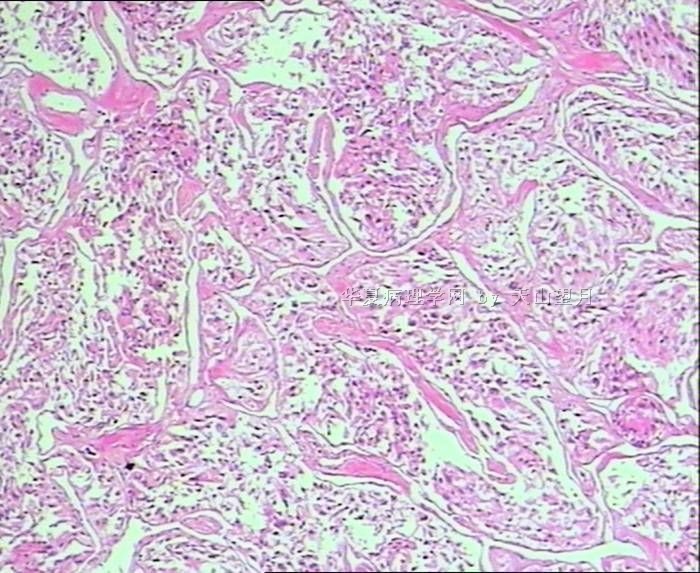
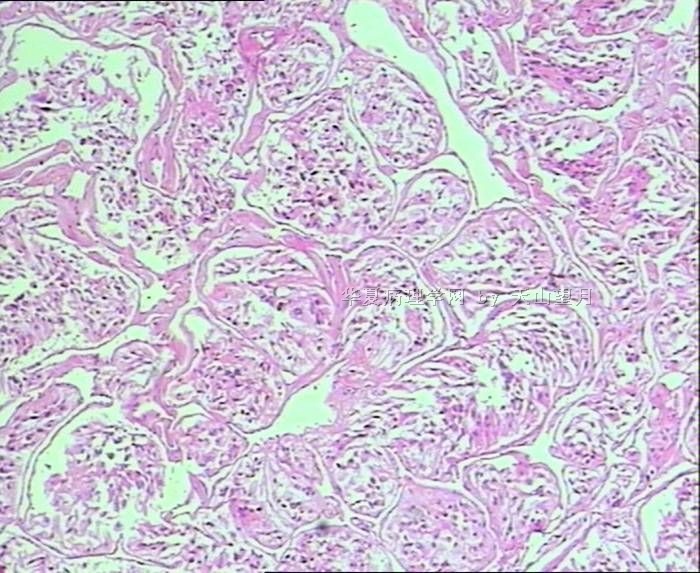
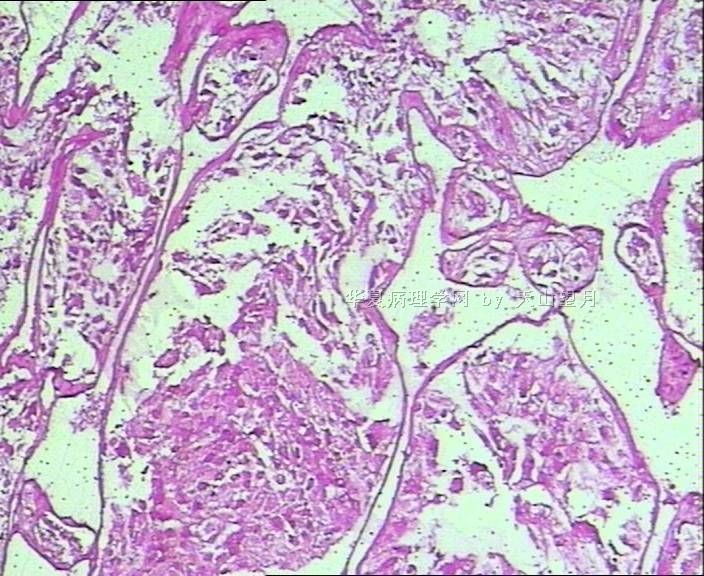
- 男,29岁,右精索肿物-新传免疫组化
-
cmuliuyang 离线

- 帖子:6
- 粉蓝豆:227
- 经验:110
- 注册时间:2010-07-01
- 加关注 | 发消息
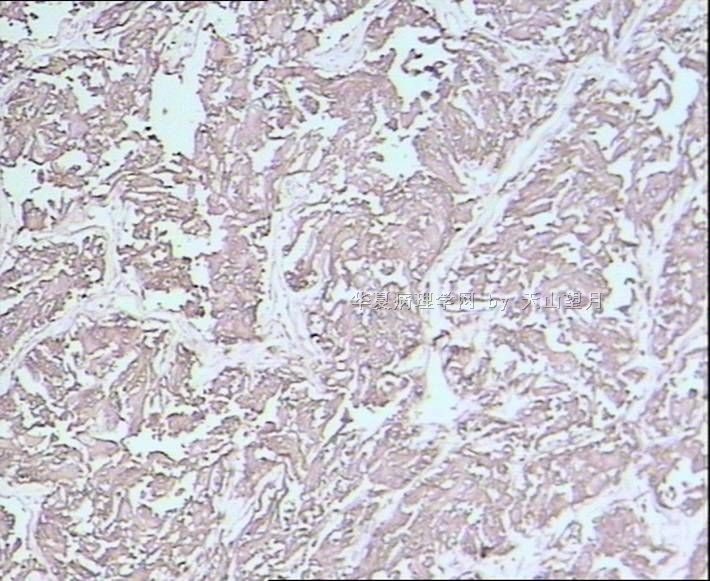
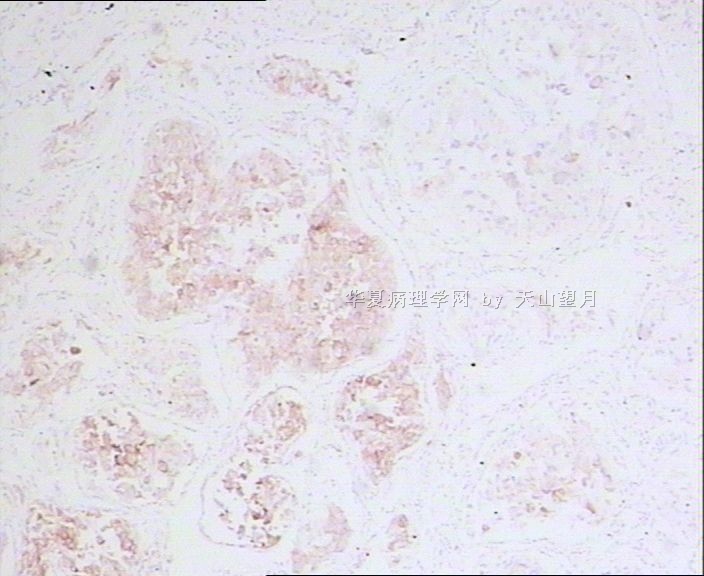
AE1/AE3、CK、EMA:标记鉴别是不是上皮性的,排除附睾管等腺样上皮成分;
PLAP、D2-40、CD117标记精原细胞,其中PLAP在恶性生精细胞呈阳性,排除小管内(原位)精原细胞瘤(在隐睾症的基础上发生);
SMA标记看有没有肌上皮可能;
Desmin、MyoD1、S-100、NSE鉴别排除腺泡状肌源性或软组织肿瘤。
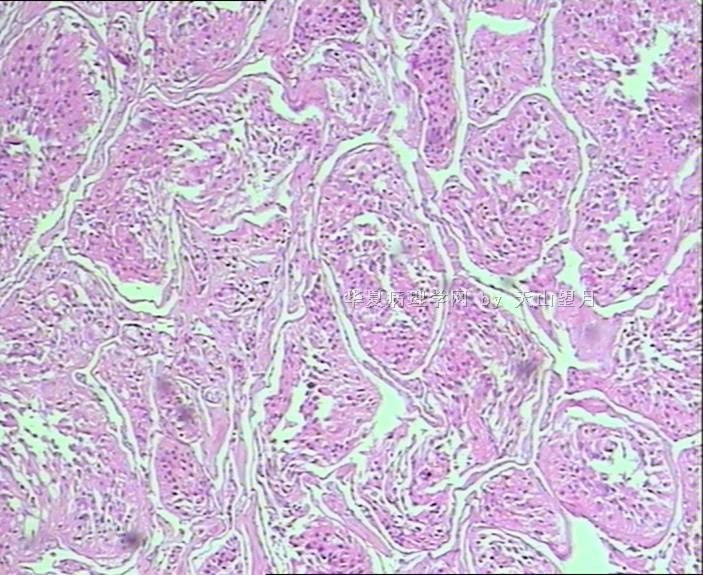
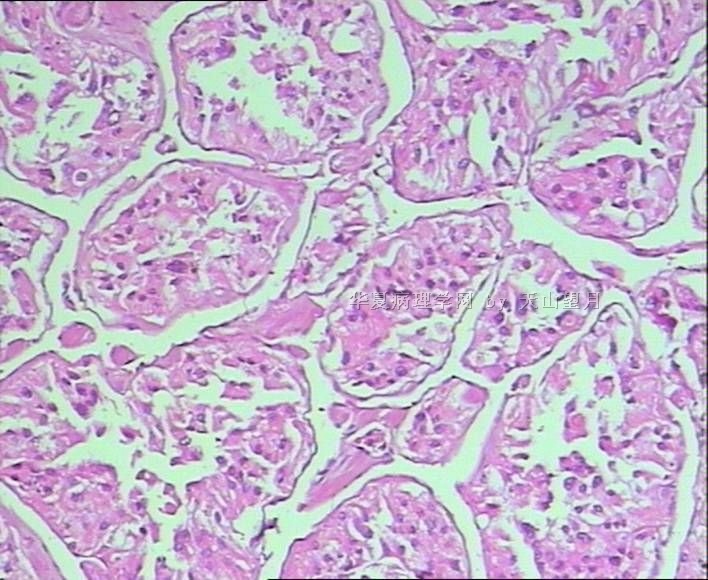
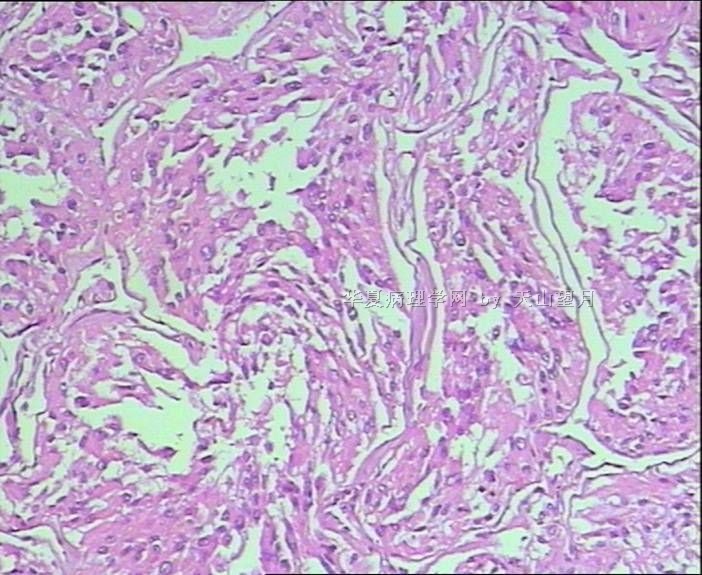
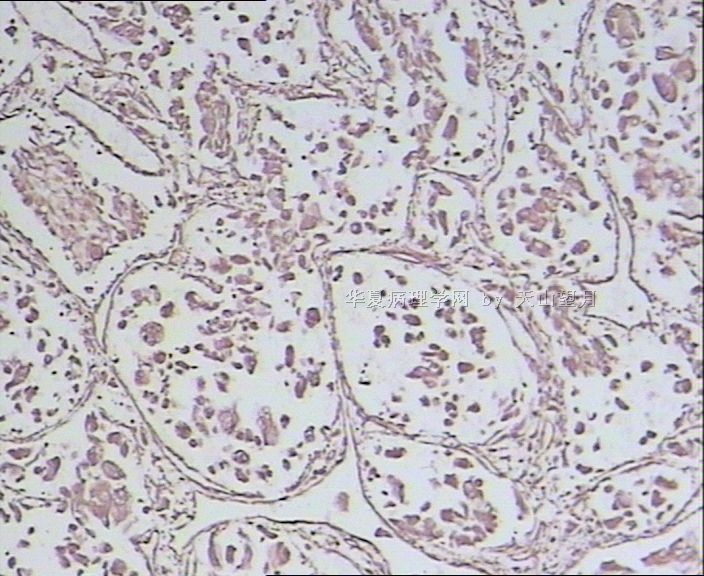
从仅有的一张图片看,倾向隐睾(如果阴囊内有睾丸缺失)或睾丸异位(如果阴囊内没有睾丸缺失,可能是因为睾丸发育及其下降过程中留下在精索或腹股沟管内的睾丸残基,发育呈异位睾丸)。由于腹腔或腹股沟管的温度不适宜于生精细胞发育与成熟,故留下支持细胞为主的曲靖小管,呈萎缩的睾丸组织。
仅供参考。

- 王军臣
-
本帖最后由 于 2010-07-05 23:59:00 编辑
请教老师,查看文献,本例的组化和形态学及PAS染色结果,结合年龄,是否应该将 Alveolar soft part sarcoma作为鉴别诊断之一?
附相关文献,请参考
问题:1、MyoD1在Alveolar soft-part sarcoma中的表达,存在争议,应当如何评价。
2、本例中的PAS结果是否可以判断为阳性。
谢谢
J Clin Pathol 2006;59:1127-1132 doi:10.1136/jcp.2005.031120
· Review
Alveolar soft-part sarcoma: a review and update
2. A T Deyrup2
+ Author Affiliations
1. 1Department of Laboratory Medicine and Pathology, Mayo Clinic Rochester, Minnesota, USA
2. 2Department of Pathology and Laboratory Medicine, Emory University Hospital, Atlanta, Georgia, USA
1. Correspondence to:
A L Folpe
Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, Minnesota, USA; folpe.andrew@mayo.edu
· Accepted 25 January 2006
Abstract
Alveolar soft-part sarcoma (ASPS) is a rare, distinctive sarcoma, typically occurring in young patients. Although it displays a relatively indolent clinical course, the ultimate prognosis is poor and is often characterised by late metastases. Recently, our understanding of the genetic events underlying the pathogenesis of ASPS has greatly increased. The historical, histopathological, ultrastructural, immunohistochemical and genetic aspects of ASPS are reviewed in this article.
Alveolar soft-part sarcoma (ASPS) is a rare, distinctive sarcoma, typically occurring in young patients. Despite a relatively indolent clinical course, the prognosis is poor and is often characterised by late metastases. Since its formal description by Christopherson et al at Memorial Sloan Kettering Cancer Center, New York, USA,1 ASPS has been the subject of considerable interest for pathologists and clinicians, owing to its unique microscopic features, uncertain line of differentiation and unpredictable clinical behaviour. In the past several years, our understanding of the genetic events underlying the pathogenesis of ASPS has greatly increased. This article reviews the historical, histopathological, ultrastructural, immunohistochemical and genetic aspects of ASPS.
HISTORICAL ASPECTS
Credit for the original description of ASPS traditionally goes to Christopherson, then a fellow in surgical pathology, working under the direction of Drs Foote and Stewart.1 In his seminal series of 12 cases taken from the archives of Memorial Sloan Kettering Cancer Center over a 17-year period, Christopherson et al coined the descriptive term “alveolar soft-part sarcoma” for a distinctive and hitherto unrecognised soft-tissue tumour. This tumour typically occurred in the extremities of young women and followed a prolonged clinical course with frequent late metastases. It was defined histologically by the presence of an organoid to pseudoalveolar pattern and large, eosinophilic tumour cells. Interestingly, Christopherson et al noted that several examples of ASPS seemed to have been previously published under a variety of other names, including “malignant myoblastoma”, “granular cell myoblastoma” and “malignant granular cell myoblastoma”.2–6 Although Christopherson et al did not describe the intracytoplasmic crystalline structures that have become one of the hallmarks of ASPS, they did quote an unpublished letter from Pierre Masson at the very end of their discussion, who noted that “trichrome stains show erythrophilic and cyanophilic inclusions. The latter have in places a crystalline structure.”1 Credit for the seminal description of these crystals thus belongs to Dr Masson, who also showed a periodic acid-Schiff stain of those crystals in his 1956 text, Tumeurs humaines: histologie, diagnostics et techniques.7
Apparently unknown to Christopherson and colleagues, ASPS had been described one year previously by Smetana and Scott in a series of 14 cases retrieved from the archives of the Armed Forces Institute of Pathology, as malignant tumours of non-chromaffin paraganglia. A careful reading of this study leaves little doubt that the cases reported by these authors were uniformly histologically and clinically identical to those later reported by Christopherson et al. Smetana and Scott chose the term malignant tumours of non-chromaffin paraganglia because the tumours resembled non-physiologically active paraganglia, postulating that primitive paraganglia-like structures may perhaps normally occur in the somatic soft tissues (a hypothesis since discredited). Smetana and Scott also independently noted the crystals of ASPS, writing that “within the cytoplasm of some of the neoplastic cells were rod-shaped, coarse, basophilic bodies of unknown nature …”.8
HISTOPATHOLOGICAL FEATURES
ASPS varies little in its appearance from case to case, and is in some respects remarkable among soft-tissue neoplasms for the absence of described variants. As originally described by Christopherson et al1 (and by Smetana and Scott8), ASPS is characterised by uniform, organoid nests of polygonal tumour cells, separated by fibrovascular septa and delicate capillary-sized vascular channels (fig 1). In these nests there is prominent cellular dyscohesion, leading to the distinctive pseudoalveolar pattern for which it is named. The organoid appearance may be completely lost and the tumour may be composed of sheets of epithelioid cells. Tumours occurring in younger patients and in confined locations, such as the tongue, often show very small nests of cells, closely mimicking true paragangliomas9 (fig 2). Intravascular tumour extension is seen in most cases (fig 3). A small minority of tumours shows unusual features such as myxoid change, cystic change, haemorrhage and a prominent lymphocytic infiltrate1,8,9,10,11,12,13,14,15,16,17,18,19,20 (fig 4).
View larger version:
Figure 1
 Low-power view of a typical alveolar soft-part sarcoma, showing an organoid, pseudoalveolar proliferation of large, eosinophilic cells and a delicate capillary network.
View larger version:
Figure 2
 Low-power view of an alveolar soft-part sarcoma with a uniform “small nest” pattern. Such cases may easily be mistaken for paragangliomas.
View larger version:
Figure 3
 Intravascular extension in an alveolar soft-part sarcoma (ASPS); this is present at the periphery of the tumour in nearly all cases when examined closely, and may account for the high rate of metastasis seen in the ASPS.
View larger version:
Figure 4
 Alveolar soft-part sarcoma with a prominent associated chronic inflammatory cell infiltrate.
The cells in the lesion have distinct borders and abundant eosinophilic to clear, somewhat granular, cytoplasm, resulting in an epithelioid appearance (fig 5). Clear cell change may be very prominent, closely mimicking renal cell carcinoma21 (fig 6). Cross striations and cytoplasmic fibrils are absent. A characteristic finding is the presence of periodic acid-Schiff-positive, diastase-resistant crystalline structures, which may be rhomboid, rod-like or spiked, in individual, sheaf-like or stacked configurations.10 These crystals can also be identified with alcian blue and trichrome histochemical stains. These crystals are not especially prominent in all cases, and multiple sections may require careful searching to identify them. ASPS cells typically have round, regular, eccentrically placed nuclei with vesicular chromatin and a prominent central nucleolus; multinucleation may be present in a few cells (fig 7). Mitotic activity is usually low and necrosis is infrequent.
View larger version:
Figure 5
 High-power view of a typical alveolar soft-part sarcoma, showing relative cellular uniformity and typical cytological features, with peripherally placed, vesicular nuclei containing a prominent central nucleolus.
View larger version:
Figure 6
 High-power view of an alveolar soft-part sarcoma, with uniform clear cell change. This is believed to be an artefact related to poor cell preservation. However, it may closely simulate metastatic renal cell carcinoma of conventional subtype.
View larger version:
Figure 7
 Multinucleation in an alveolar soft-part sarcoma, a relatively frequent finding.
Although this description applies to most ASPSs, rare cases may deviate from a typical ASPS by virtue of the presence of anaplastic-appearing tumour cells with marked variation in nuclear size.1,15,20 As noted by Evans,20 these anaplastic-appearing foci may also show other atypical features, such as sheet-like growth, frequent mitotic figures and coagulative tumour cell necrosis. Extremely rare cases with spindling and xanthomatous change have also been described.15,22
Ultrastructural features
The first ultrastructural study on ASPS was that by Shipkey et al10 in 1964, with subsequent important studies over the next 25 years by Font et al,23 DeSchryver-Kecskemeti et al,24 Mukai et al25 and Ordonez et al,12 among others. ASPS cells typically rest on an incomplete basement membrane and contain only rare, primitive cell–cell junctions. The cytoplasm contains numerous mitochondria and an extensive Golgi complex with adjacent small dense granules.10,12,23–25 The endoplasmic reticulum is usually, but not always, sparse. The distinctive crystals of ASPS are typically found intermingled with these dense granules. They are often bounded by a single membrane and are composed of a periodic latticework of fibres (diameter 4.5–5 nm and periodicity 10 nm; fig 8).10,12,23–25 The composition of these crystals was interpreted as a product similar to renin24 or aggregates of actin filaments,25 and it was suggested that they could be similar to the inclusions seen in rhabdomyoma or nemaline rod myopathy.26 However, elegant recent work by Ladanyi et al,27 including ultrastructural immunohistochemistry, has convincingly shown that the crystals consist of aggregates of the monocarboxylate transporter protein MCT1 and its cellular chaperone CD147.27
View larger version:
Figure 8
 Electron micrograph, showing a typical intracytoplasmic crystal in an alveolar soft-part sarcoma (reproduced with the permission of Dr Cyril Fisher, The Royal Marsden NHS Trust, London, UK).
IMMUNOHISTOCHEMICAL FINDINGS
Until recently, immunohistochemical analysis played a limited part in the diagnosis of ASPS. Several studies have examined ASPS immunohistochemically, both with and without the use of epitope-retrieval techniques, often with differing results (reviewed by Foschini and Eusebi28). In general, ASPSs are negative for epithelial markers, such as cytokeratins and epithelial membrane antigen, negative for specific neuroendocrine markers such as chromogranin A and synaptophysin, and negative for specific melanocytic markers, such as HMB45 and Melan-A. Non-specific markers such as neurone-specific enolase and vimentin may be present in roughly 30–50% of cases. There has been a great deal of interest in the expression of muscle-related proteins in ASPS, due to the prevailing theory for many years that ASPS represented an unusual form of myogenic tumour. Antibodies to actins in pan, smooth and skeletal muscle have been reported to be positive in nearly 50% of patients with ASPS. Actin expression in pan and smooth muscle is by no means specific for myogenous differentiation, and the ASR-1 antibody to skeletal-muscle actins is notoriously difficult to interpret because of high levels of non-specific staining. Desmin expression has been reported in around 50% of cases with ASPS, although it tends to be present in only a small number of the neoplastic cells. Again, it should be emphasised that desmin expression is not limited to myogenous tumours and can be seen in a wide variety of other lesions, including melanoma, tenosynovial giant-cell tumour, Ewing’s sarcoma and angiomatoid “malignant” fibrous histiocytoma, among others.21
A much more controversial topic has been whether ASPS expresses truly specific markers of skeletal muscle differentiation, such as the myogenic nuclear regulatory proteins MyoD1 and myogenin. In 1991, Rosai et al29 reported a case of ASPS that seemed to show convincing MyoD1 expression by immunofluorescence and western blotting on fresh-frozen tissue.29 This was followed by Tallini et al’s30 report on a MyoD1-positive ASPS, using immunohistochemistry on frozen sections. Regrettably, these two early findings have not been substantiated by any subsequent study. Three separate studies, with commercially available antibodies and modern immunohistochemical techniques, including heat-induced epitope retrieval, have not identified expression of either MyoD1 or myogenin in the more than 35 cases studied.31–33
It has recently been discovered that ASPSs are characterised by a tumour-specific der(17)t(X;17) (p11;q25) that fuses the transcription factor 3 (TFE3) gene at Xp11 to the ASPL gene at 17q25, creating an ASPL–TFE3 fusion protein.34 Recently, an antibody directed against the C-terminus of the TFE3 has emerged as a highly sensitive and specific marker of the ASPS35 (fig 9). Although TFE3 seems to be almost universally expressed in normal tissues, this expression is at very low levels and strong nuclear expression of TFE3 is seen almost exclusively in tumours known to contain the TFE3 gene fusions, such as ASPSs and rare paediatric renal carcinomas.35 It must be emphasised that only nuclear expression of TFE3 is of diagnostic value, as cytoplasmic staining (possibly non-specific) is seen in various tumours. The TFE3 antibody binds to the protein downstream of exon 4 and therefore is positive in both fusion variants seen in ASPS. Importantly, however, strong TFE3 is seen in granular cell tumours,35 a potentially confusing and somewhat ironic finding, given the original classification of ASPS as a “malignant granular cell myoblastoma” (malignant granular cell tumour).
View larger version:
Figure 9
 An alveolar soft-part sarcoma (ASPS) showing uniform, strong nuclear positivity with an antibody directed against the C-terminus of transcription factor 3 (TFE3), confirming the presence of an ASPL–TFE3 fusion protein.
CYTOGENETIC AND MOLECULAR GENETIC FEATURES
The earliest cytogenetic analysis of ASPS identified multiple structural and numerical abnormalities, strongly suggesting a role for chromosomal abnormalities in the pathogenesis of this tumour.36 Subsequent karyotypes identified translocations between chromosomes X and 1737–41 and, in 1998, both break points were identified: 17q25 and Xp11.2.42 Subsequently, Ladanyi et al34 characterised the two genes concerned in the translocation.
ASPS is characterised by an unbalanced translocation: der(17)t(X:17)(p11;p25). This translocation results in the fusion of a gene of unknown function, ASPL, on chromosome 17 to the TFE3 gene on the X chromosome.34 Consequently, the ASPL gene is joined in frame upstream of either the third or fourth exon of TFE3, yielding two fusion variants, type 1 and type 2, respectively (fig 10). Usually, fusion genes are generated by reciprocal balanced translocations with only one of the two chimeric genes being pathogenic. The translocation in ASPS is unusual in that it is unbalanced, although two patients with a reciprocal translocation have been described.34,43 It has been suggested that the female predominance seen in patients with ASPS is because of the presence of two X chromosomes in these patients, increasing their chances of a translocation on this chromosome.34
View larger version:
Figure 10
 Structure of the alveolar soft-part sarcoma (ASPL), locus gene transcription factor 3 (TFE3) gene and the ASPL–TFE3 fusion gene. AD, activation domain; bHLH, basic helix–loop–helix; LZ, leucine zipper; UT, uridine triphosphate.
ASPL is a ubiquitously expressed protein with greatest expression in adult heart, skeletal muscle, pancreas and testis, and somewhat lower expression in fetal tissues.34 Although little is known about the ASPL protein, the gene itself has been partially characterised: the cDNA is 1872 nucleotides in length, yielding a predicted protein of 476 amino acids. Sequence comparison with known human genes has not identified major homology; however, there is some similarity to sequences from other species, including yeast, fruit fly and worm;34 the protein function is unknown in all of those species. At the structural level, a sequence homologous to the UBX motif was found in the C-terminus of ASPL.34 The UBX domain is an 80-amino acid module localised in the C-terminus of several eukaryotic proteins and is hypothesised to have a role in ubiquitin-related processes.44 No other complex domains have been identified in the ASPL sequence.
TFE3 is much more understood: it is one of a family of basic helix–loop–helix leucine zipper transcription factors that includes MiTF, TFEB and TFEC.45 They are ubiquitously expressed and bind DNA as homodimers and heterodimers. All possible pairwise combinations have been identified in vitro, and it has been suggested that tissue-specific gene expression is regulated by the relative coexpression of each protein.45 Two activation domains have been identified in TFE3: an acidic, N-terminal domain and a proline-rich C-terminal domain.46 These two domains act synergistically and differential splicing provides additional regulatory control. The loss of the N-terminal-activating domain, as is seen in fusion variant 1, results in a dominant negative form of the transcriptional activator that interferes with full-length TFE3.47
The composite ASPL–TFE3 protein loses the ASPL UBX domain but retains the TFE3 basic helix–loop–helix DNA-binding domain, leucine zipper dimerisation domain and C-terminal activation domain. The ubiquitous expression of ASPL suggests constitutive expression of the ASPL promoter and, accordingly, it has been hypothesised that the loss of the TFE3 upstream regulatory elements may contribute to the pathogenesis of this translocation.34 In the type 2 fusion, the N-terminal TFE3-activating domain is lost and, although no clinical difference has been identified between the two fusion variants, only a few cases have been analysed.34,43
Interestingly, TFE3 is partnered with several other genes in balanced reciprocal translocations in a subset of (usually) paediatric papillary renal cell adenocarcinomas.48–52 In these tumours, several possible translocations can be seen: (1) the entire coding region of TFE3 is fused with the novel gene PRCC at 1q21.2 or (2) TFE3 minus the first three exons is fused to one of two splicing factor genes, PSF at 1p34 or NonO at Xq12. The second case is analogous to that seen in ASPL–TFE3 type 1 fusion as, in both instances, the TFE3 activation domain is lost, whereas the basic helix–loop–helix DNA-binding domain is retained.
CLINICAL FEATURES AND BEHAVIOUR
ASPS is extremely rare, and is generally believed to account for <1% of soft-tissue tumours overall.21 ASPS has, however, accounted for roughly 10% of cases reported from large sarcoma referral centres, in both adults18 and children.14 As many as 60% of ASPS cases are seen in women,18 although this female sex predilection may be less pronounced in children.14,17 The tumour typically occurs in the deep soft tissues, most often in the buttock and thigh, with a smaller number of cases at other soft-tissue locations such as the arm, chest and retroperitoneal tissues.1,8,9,10,11,12,13,14,15,16,17,18,19,20 In children, a substantial percentage of cases occur in the head and neck, often in the orbit23 or tongue.9 ASPSs have been reported in a wide variety of locations, including the bladder,6 stomach,53 gynaecological tract54 and bone.55 Most often, ASPSs present as painless masses, which may be highly vascular on imaging studies.21
ASPS behaves as a relatively indolent, but relentless sarcoma, characterised by late metastases and an extended clinical course; survival rates of 77% at 2 years, 60% at 5 years, 38% at 10 years and only 15% at 20 years have been reported by Lieberman et al.18 Studies on another large series from the MD Anderson Cancer Center, Texas, USA, found a 5-year disease-free survival of 71% in patients presenting with localised disease as compared with only 20% in patients presenting with metastases.56 The prognosis for children with ASPS may be considerably better, with Pappo et al14 reporting disease-free survival in 9 of 11 cases (with a relatively short follow-up duration) and Casanova et al17 reporting 100% survival at >5-year follow-up in 12 patients with localised disease at presentation. Lingual and orbital tumours also have very high survival rates, possibly reflecting a combination of small size at the time of diagnosis and younger patient age.9,23
No histopathological features are predictive of prognosis in patients with ASPS, and these tumours should not be graded under either the National Cancer Institute or French grading schemes. NCI57 Features associated with improved prognosis include younger age at diagnosis, small tumour size and the presence of localised disease.18,56 ASPSs most often metastasise to lungs, brain and bone. Metastases, sometimes to unusual locations such as the breast, may be the presenting symptom in some patients with ASPS. Adjuvant chemotherapy does not seem to be effective in the treatment of ASPSs, although there may be some role for adjuvant radiotherapy in reducing the risk for local recurrences.18,56,58
LINE OF DIFFERENTIATION
The cell of origin or, better, line of differentiation taken by ASPS has been the subject of considerable speculation and controversy over the past 50 years. ASPS was originally regarded as a malignant variant of granular cell myoblastoma, a term that encompasses the entity now known as granular cell tumour.2–6,57,59,60 The speculation that ASPSs represented unusual malignant tumours of non-chromaffin paraganglia8 has subsequently been disproved by several ultrastructural, histochemical and immunohistochemical studies, documenting the complete absence of neuroendocrine differentiation in ASPS.25,61 Similarly, the suggestion that ASPSs were malignant angioreninomas has been disproved by negative immunohistochemical studies for renin, as well as by the absence of clinical signs of hyper-reninism in patients with ASPS.25 Perhaps the longest lived hypothesis has been that ASPS is an unusual myogenous tumour, on the basis of some ultrastructural similarity of a component of the ASPS crystal to actin filaments,25 overlap between the appearance of these crystals and those seen in nemaline myopathy and rhabdomyoma,26 and variable expression of muscle-related proteins, such as actins and desmin.28 However, as discussed in the section Immunohistochemical findings, the specificity and significance of these immunohistochemical findings have been questioned, and there is no convincing evidence of expression of truly muscle-specific proteins in ASPS.62
In the light of the recent discovery that ASPS is part of the family of translocation-associated sarcomas, there exists an additional hypothesis—that ASPS displays a scrambled phenotype, without a normal counterpart.63 This would seem to be a compelling hypothesis, given the lack of resemblance of ASPS to any known structure, its resolute defiance of all attempts to categorise it and our increasing understanding that a subset of human neoplasms, such as perivascular epithelioid cell neoplasms, possibly lack normal counterparts.57 Alternatively, it cannot be entirely excluded that ASPS recapitulates the phenotype of a yet-to-be discovered, extremely rare, normal cell of unknown function.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of ASPS is fairly broad, and revolves around neoplasms that may show nested or organoid patterns of growth and cells with abundant eosinophilic cytoplasm. Renal cell carcinomas, adrenal cortical carcinomas and hepatocellular carcinomas may mimic ASPS by virtue of their abundant eosinophilic to clear cytoplasm. Immunohistochemical evidence of strong cytokeratin expression, expression of site-associated markers (eg, renal cell carcinoma antigen in renal cell carcinoma, Melan-A crossreactivity in adrenal cortical carcinoma and HepPar1 in hepatocellular carcinoma) and absence of TFE3 expression should help in this differential diagnosis. Paediatric renal cell carcinomas with TFE3 gene fusions may show cytokeratin expression and TFE3 expression to be absent; clinical correlation as to the presence of a renal mass is critical in this situation. Malignant melanoma may simulate ASPS, but should be easily separated immunohistochemically for S100 protein, HMB45 and Melan A. Paragangliomas, unlike ASPSs, show strong expression of chromogranin A and synaptophysin. Alveolar rhabdomyosarcoma, despite its somewhat similar name, appears as an entirely different “small blue round cell tumour”, which strongly expresses desmin and myogenin nuclear regulatory proteins. Perivascular epithelioid cell neoplasms, such as epithelioid angiomyolipoma, coexpress smooth-muscle actin and melanocytic markers, and are almost always TFE3-negative. In most instances, granular cell tumours lack the striking cytological atypia seen in ASPS and show strong S100 protein expression. TFE3, however, may be expressed by granular cell tumours, a potential pitfall.
CONCLUSIONS
ASPSs are rare soft-tissue sarcomas with a distinctive and generally invariable histological appearance. It is now clear that they are caused by a specific unbalanced translocation, der(17)t(X:17)(p11;p25), which results in the formation of an ASPL–TFE3 fusion gene. The presence of this fusion gene can be detected by either molecular genetics or immunohistochemical analysis for the C-terminus of the ASPL–TFE3 fusion protein, assisting in the diagnosis of ASPS and in its distinction from the wide variety of other neoplasms with which it may be confused. The presence of this unique fusion gene also suggests that ASPS lacks a normal cellular counterpart.
谢谢Dr.didnx提供详细文献。
腺泡状软组织肉瘤(ASPS)好发于青少年,30以下者多见于女性,30岁以上者,多见于男性。如发生在精索实属罕见。组织结构呈器官样或腺泡样排列,瘤细胞核大,空泡状,大核仁,核分裂和坏死少见,胞浆透明,PAS染色有70-80%显示PAS阳性棒状(或针状)结晶。IHC标记可表达肌源性标志物,如50%左右病例Desmin阳性,部分病例SMA和MyoD1阳性,但MyoD1表达是在胞浆中,而不是在胞核(有别于腺泡状横纹肌肉瘤);可表达S-100和NSE(但无意义),不表达myogenin。
腺泡状横纹肌肉瘤(ARMS)在任何年龄均可发生,但好发于年龄偏小的青少年。多见于四肢深部,也可发生在躯干和会阴部等。经典型的组织结构也为腺泡状,但细胞原始——小而胞质少,核染色深,核分裂多见,有坏死和细胞脱落于假腺内,总能在局灶见到胞浆嗜酸性和横纹肌母细胞分化。IHC标记显示表达横纹肌表型,Myogenni表达并分布于腺泡或血管周围呈强表达。
所以,本例要鉴别ASPS和ARMS,需要根据组织结构、细胞形态和免疫表型(特别是MyoD1的定位以及有无Myogenin表达)的仔细推敲。
- 王军臣











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}