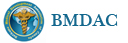
| 图片: | |
|---|---|
| 名称: | |
| 描述: | |
- 腹腔肿块
-
本帖最后由 于 2010-05-27 21:38:00 编辑
| 以下是引用c7514975在2010-5-27 21:22:00的发言: 促纤维组织增生性小细胞肿瘤 |
促纤维组织增生性小细胞肿瘤(Desmoplastic Small Round Cell Tumor,DSRCT)没有肌上皮的表达。
促纤维组织增生性小细胞肿瘤(desmoplastic small round cell tumor, DSRCT)也叫硬化性小细胞肿瘤是近年才被确认的一种少见的恶性肿瘤。此肿瘤由Gerald和Rosai于1989年首先报道,其特点为沿浆膜侵袭和播散性生长,由呈巢性小细胞和硬化性间质构成的组织结构,以及免疫组化有特殊的上皮、间叶和神经性标记物的复合表达。最初文献报道的名称不同,包括:伴有异向分化的硬化性小细胞瘤、腹腔内硬化性小圆细胞瘤、表达间叶型中间丝的腹膜恶性小细胞上皮性肿瘤、伴异向分化的腹膜硬化性小圆细胞瘤、伴有异向分化的儿童腹腔神经外胚瘤和伴多向分化的硬化性小细胞瘤等。尽管DSRCT发生得较少,文献报道仅几百例,但近2-3年来对它的研究却相当深入和广泛(11篇文献),从临床与治疗、病理组织学形态、超微结构到基因及免疫表型等,在各个方面都得到了很好的认识。现己证实此肿瘤具有独特的临床、细织、免疫细织化学和细胞基因特征。
1 临床表现
DSRCT主要发生在青年男性,年龄最小3岁,最大52岁,平均年龄在18-25岁之间,男女之比为4-5:l。大多数肿瘤发生在腹腔内,最常见的临床症状是病人出现腹部疼痛性包块,在疾病的早期常常伴有便秘,个别患者发生肠梗阻、部分患者最初症状与尿道受压有关,如排尿困难。多数患者腹部可触摸到包块,常有腹水。其X线常表现为“间皮瘤样”生长方式,肿瘤沿腹腔表面播散性生长。手术所见与X线表现相同,常发现腹腔中有较大的肿块,一般位于盆腔或网膜上,不直接与任何腹腔器官发生联系,肿物广泛在腹腔和盆腔的腹膜上种植,如同不知道原发灶的多结节性累及腹膜的转移性疾病。淋巴结有时也可以受累,一些病例肿瘤也可浸润到胃、小肠、肝、胰、膀胱、脾或卵巢。几乎所有病例肿瘤都难以被完全切除。
发生在其它部位的DSRCT也有文献报道,包括胸腔、中枢神经系统、阴囊、附睾、卵巢、肝脏及手臂等。起源于胸腔内病变的患者常有胸痛,X线和临床表现与间皮瘤相似,并可累及纵隔和包绕食道,l例发生于脑脊膜和脊髓内肿块的病例,患者出现头疼、呕吐和眩晕症状。最近有人报道了6例附睾区域的DSRCT,在阴囊、附睾或睾丸出现疼痛性或无痛性包块,其中3例同时存在远处转移(颈部及腹膜后淋巳结,肺)。另外,少数病人是以颈部和腋下淋巴结转移为首发症状,l例报道有肝脏肿瘤伴双肺转移,但无腹腔受累倩况。
2大体形态
肿瘤由多结节组成,一般是一个较大的肿瘤伴同许多小瘤结,所报道的肿瘤最大直径达40Cm,重2000g,肿物表面光滑、呈圆凸形、切面为实性、灰白色,有时可见到界限清楚的坏死和出血区,一些病例出现假囊性和粘液样区域、有1例报道肿瘤带蒂,呈多囊性并含血液。
3 组织学改变及类型
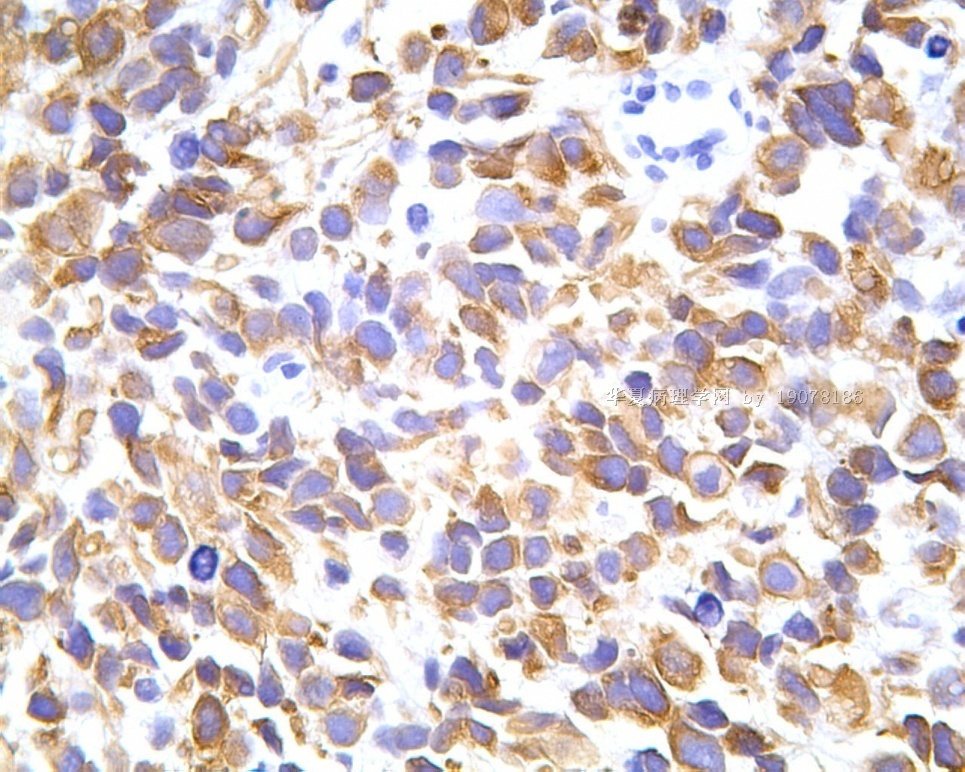
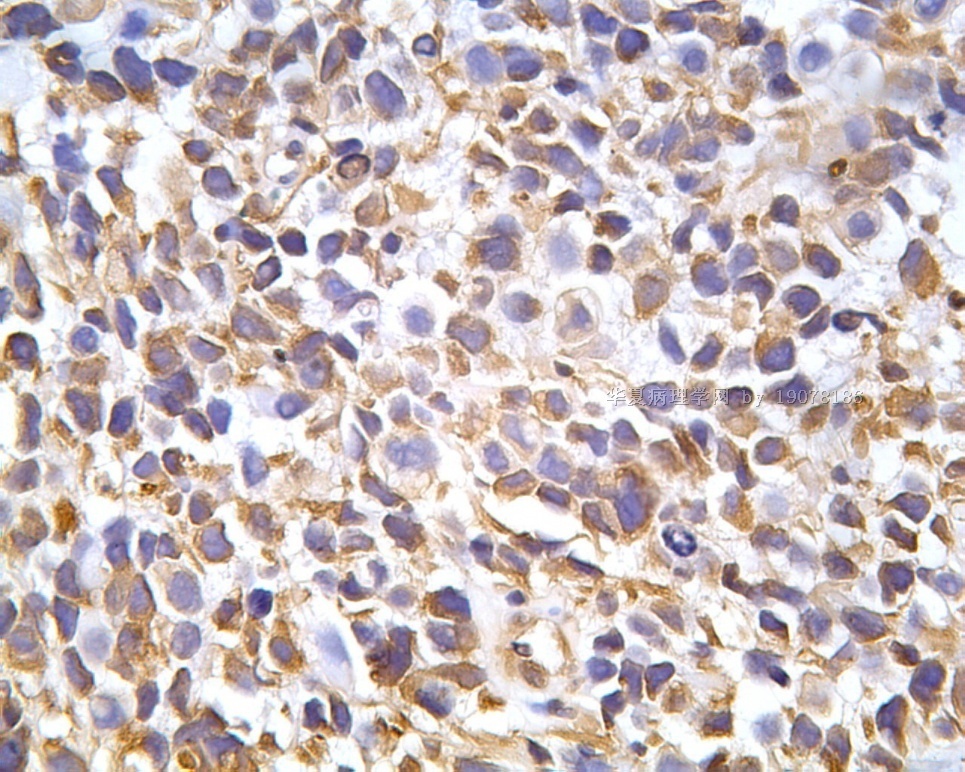
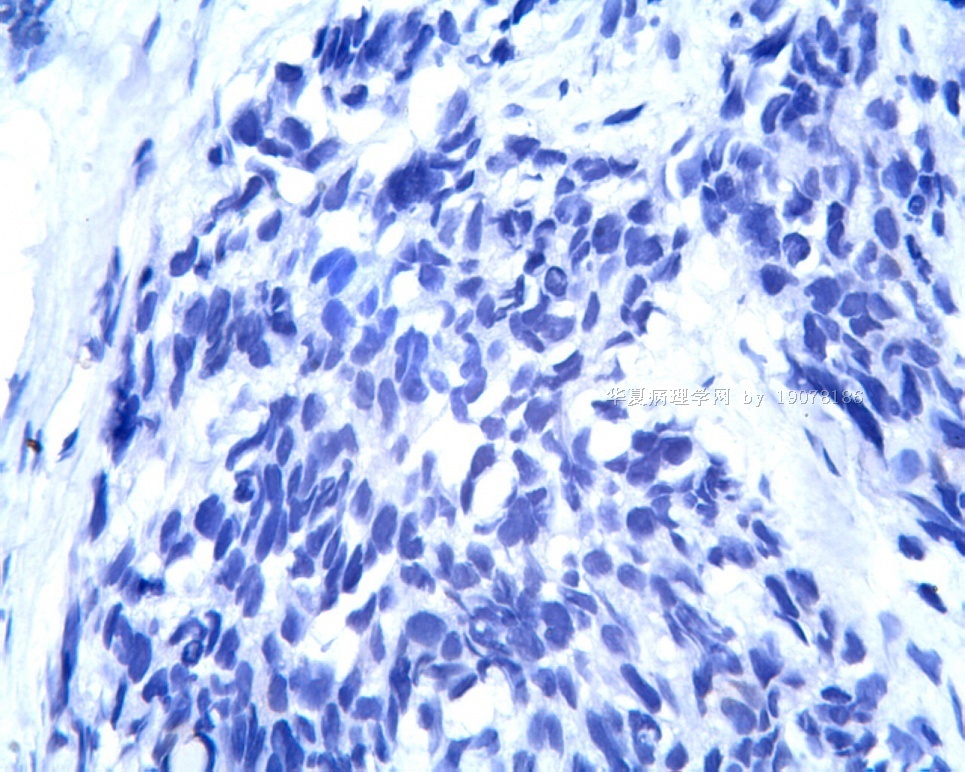
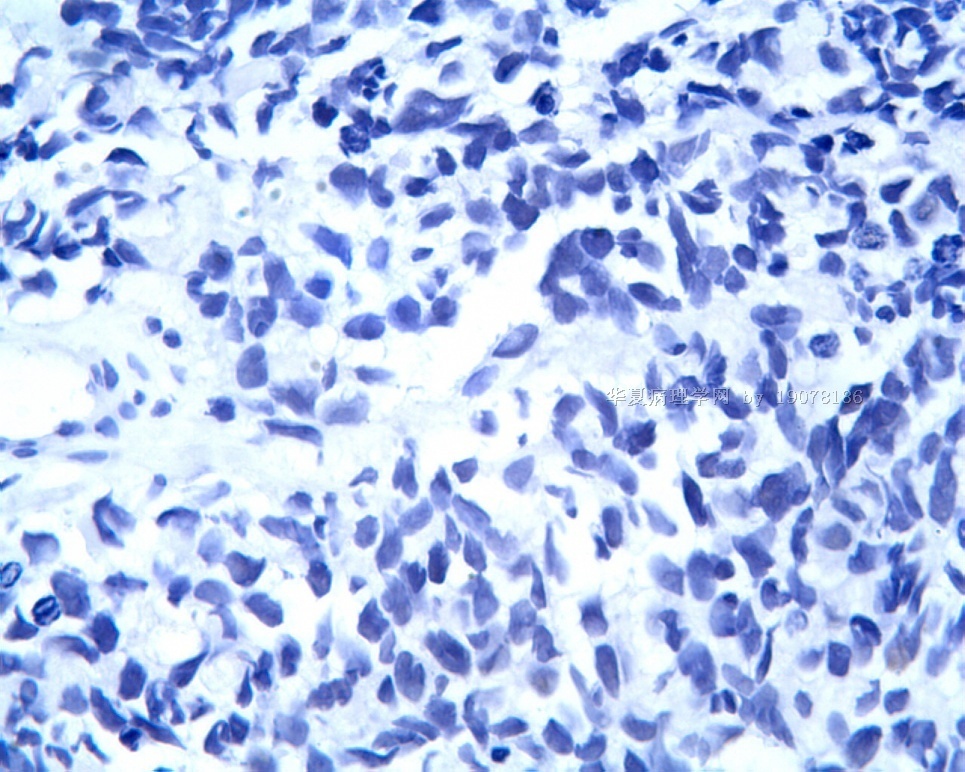
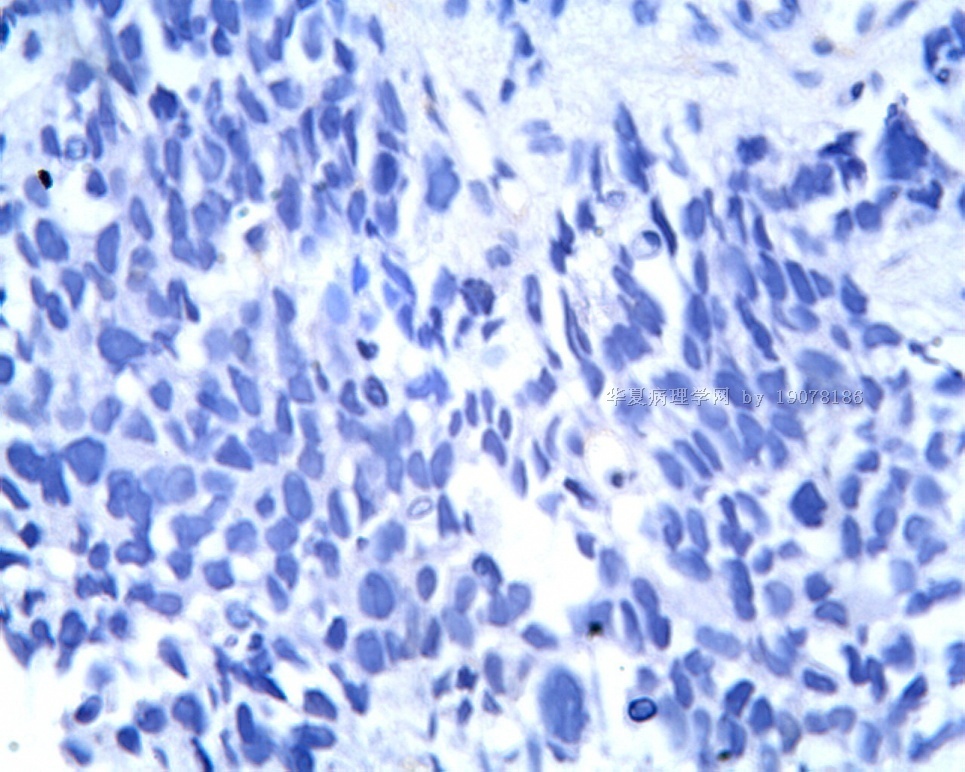
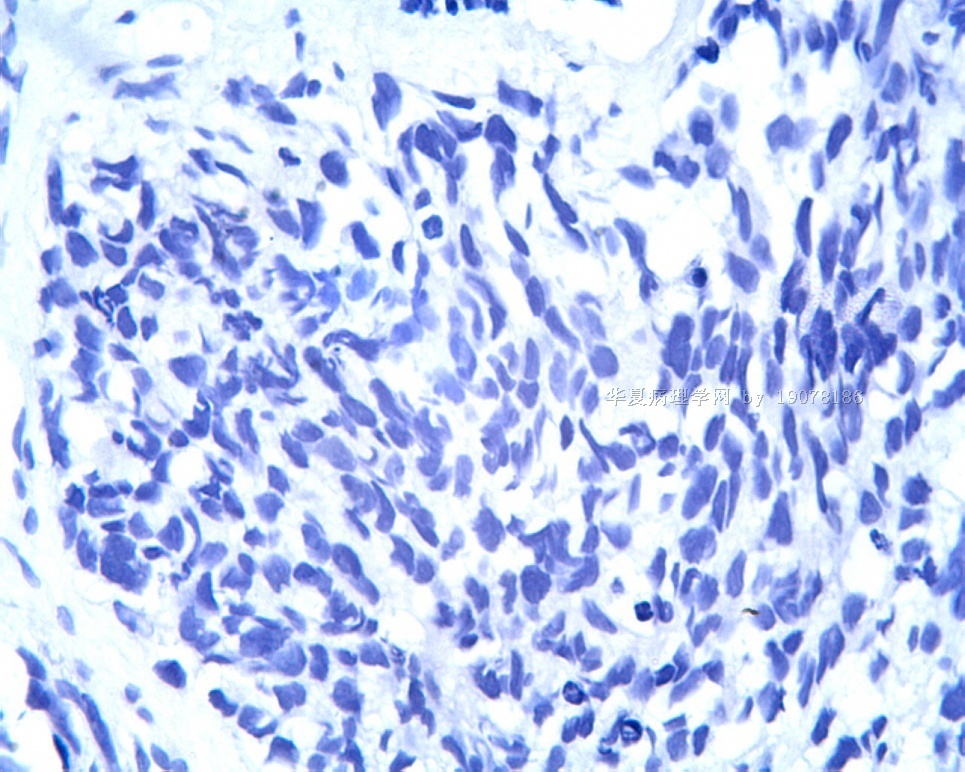
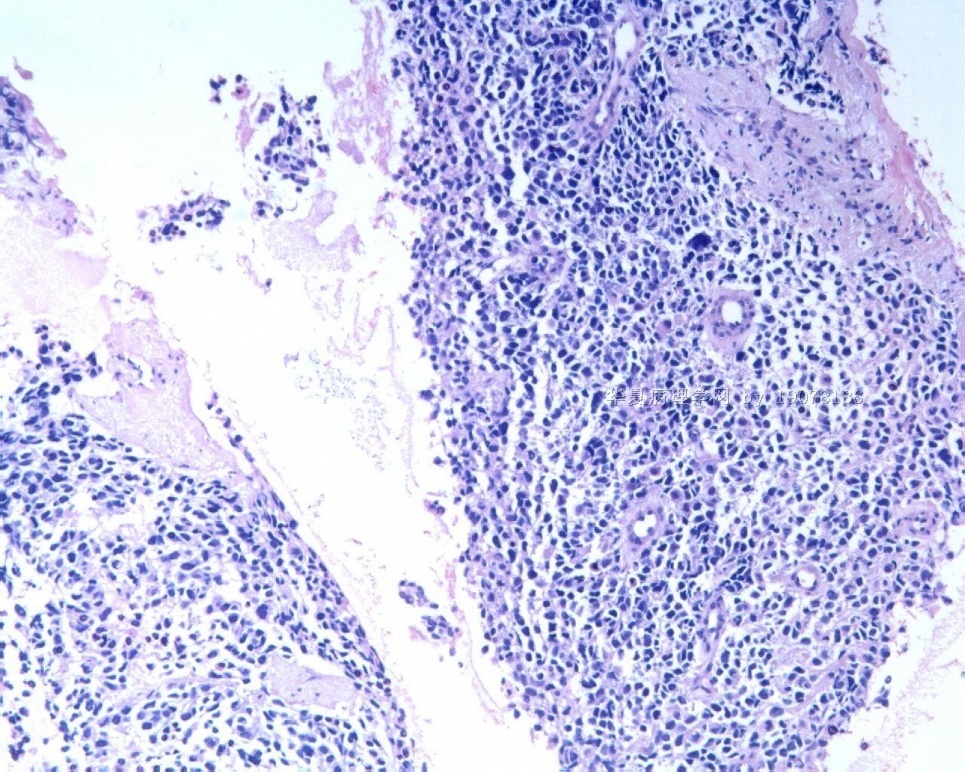
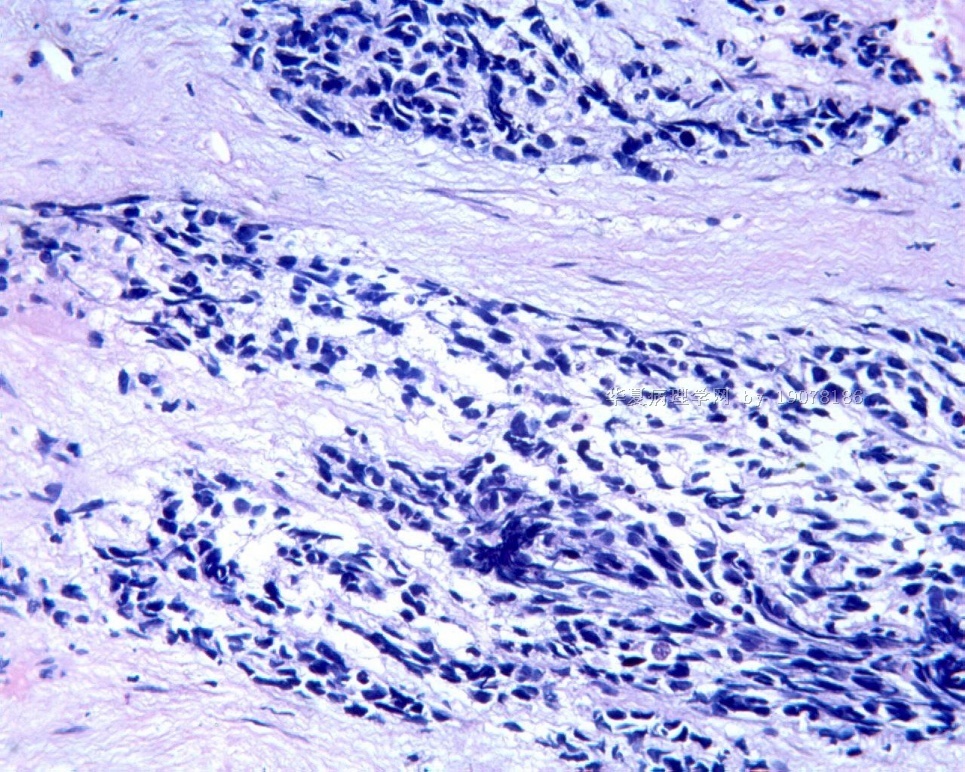
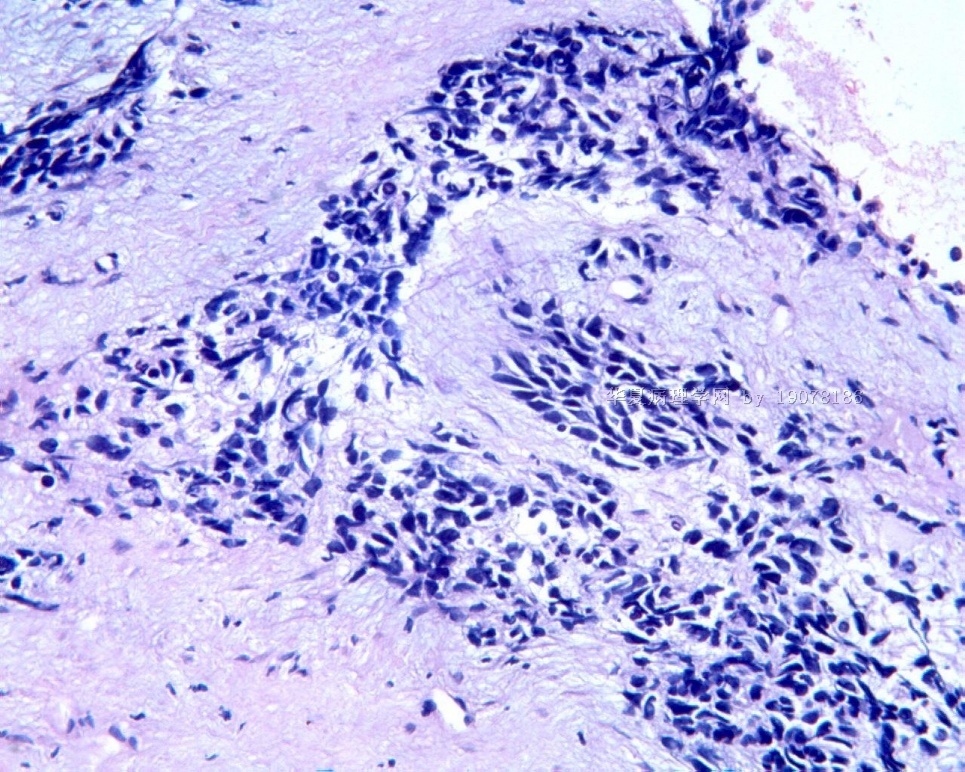
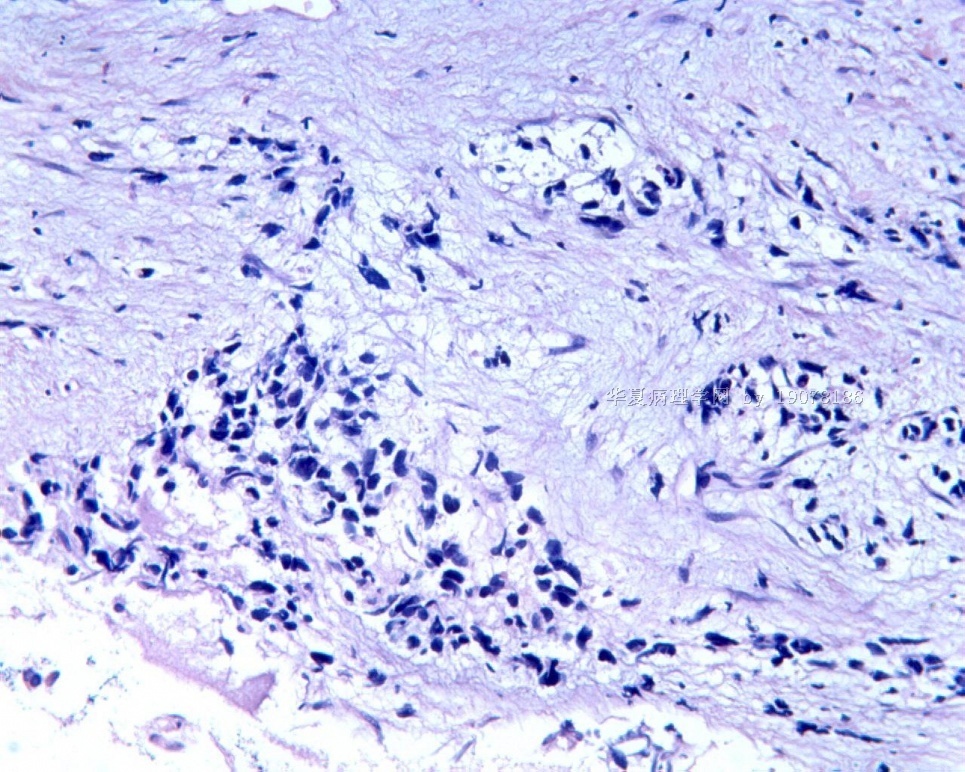
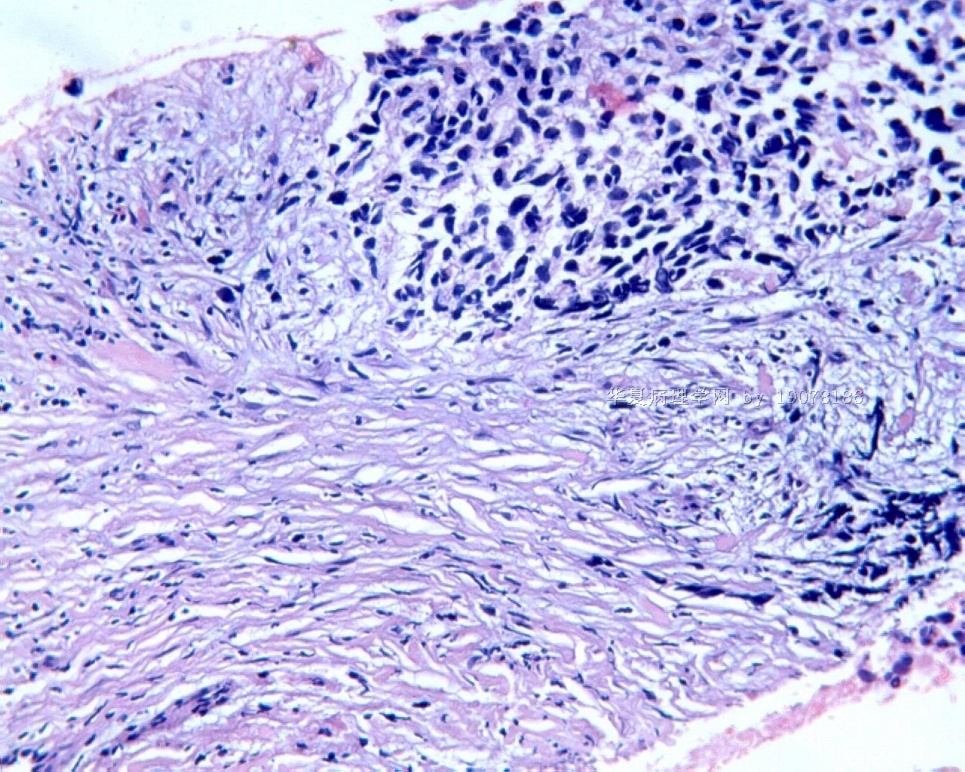
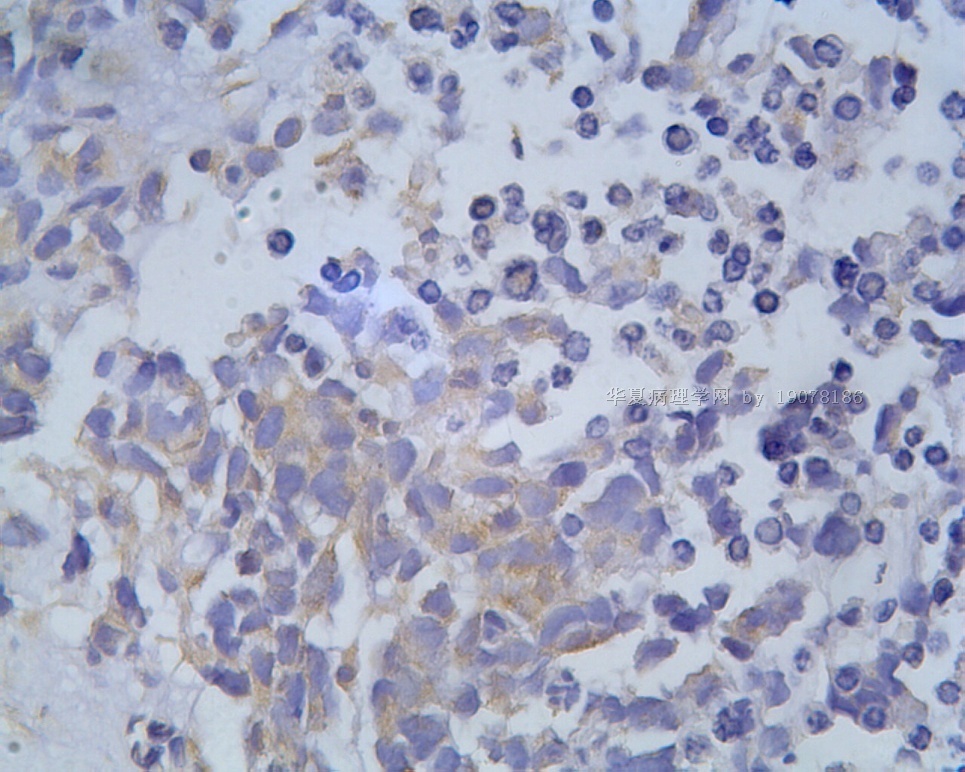
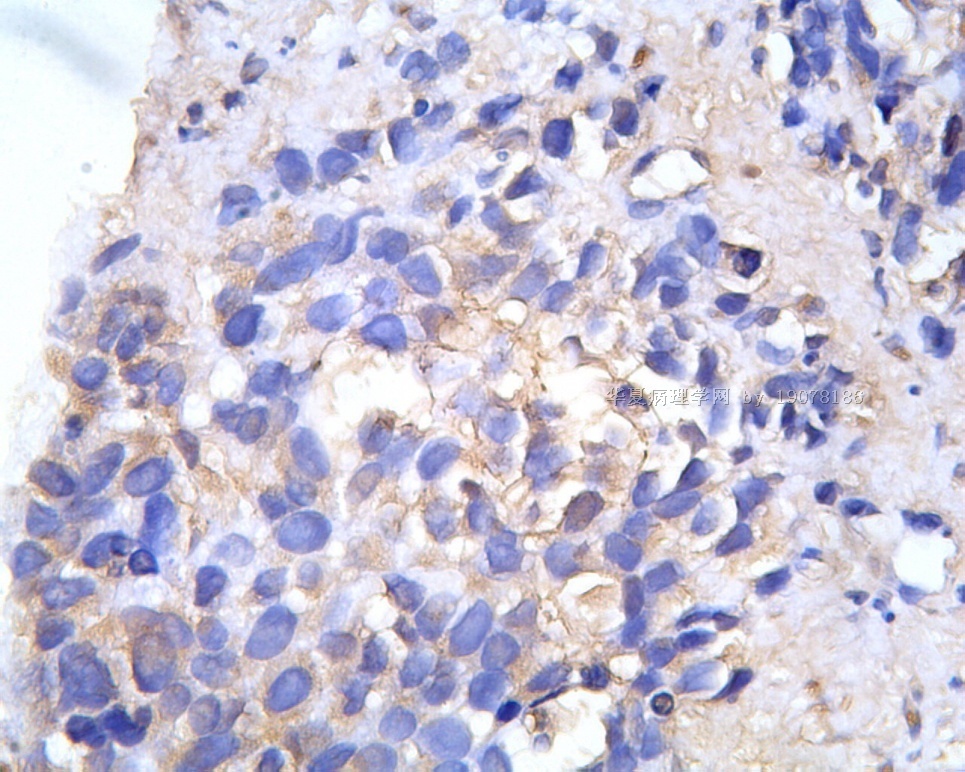
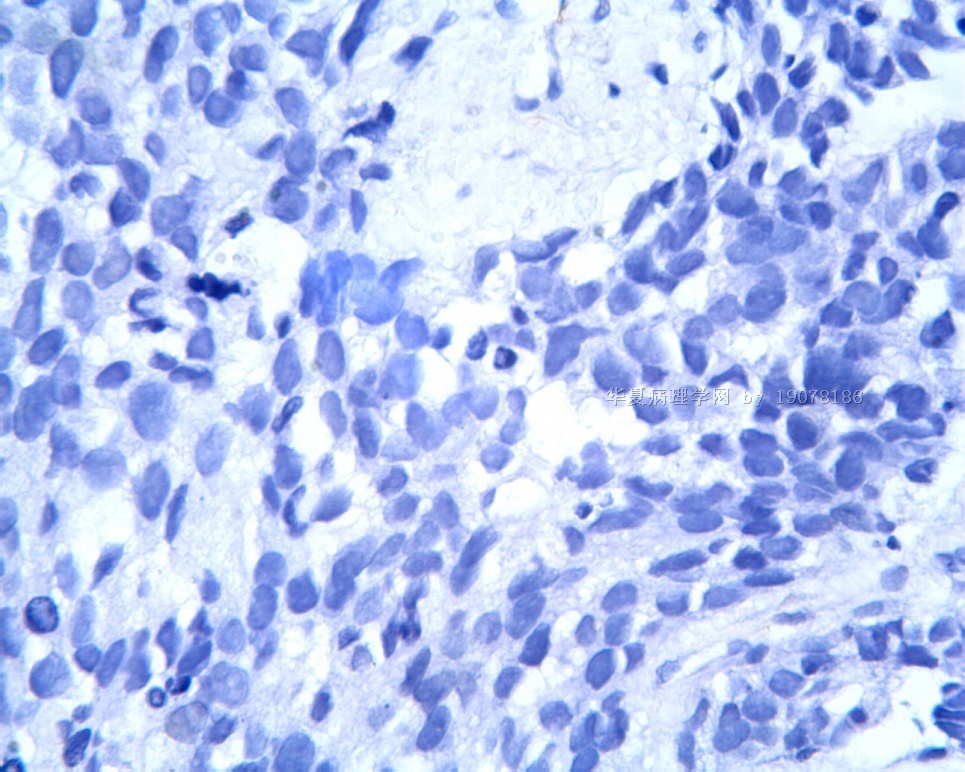
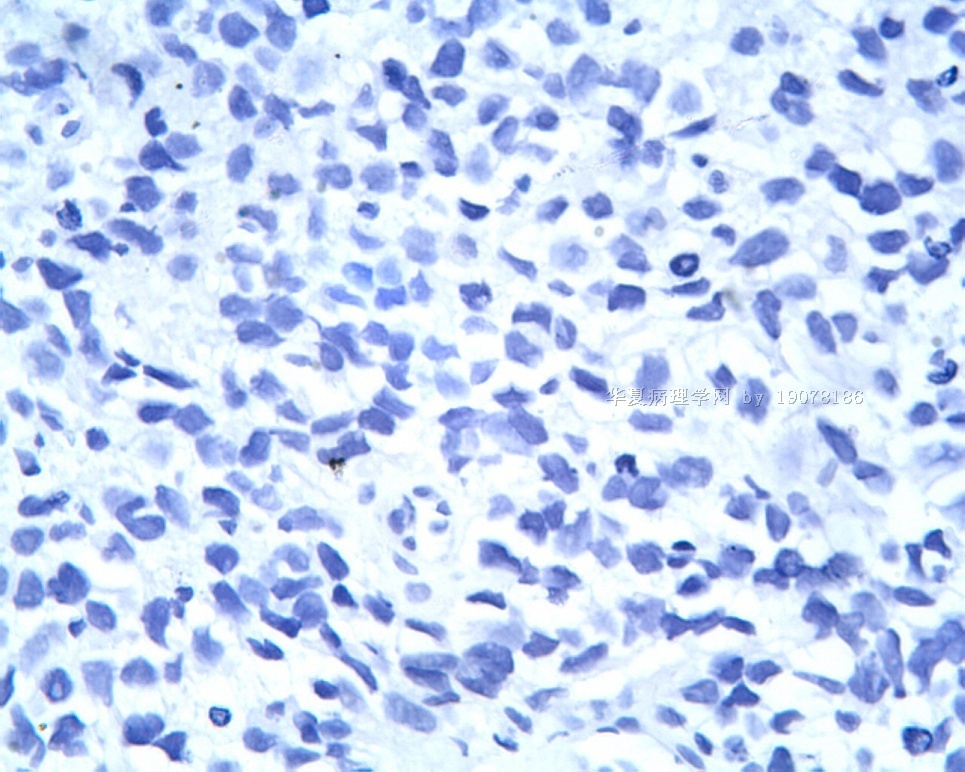
3.1 DSRCT典型的组织学改变 低倍镜下见肿瘤细胞小而圆,形成无规则的但轮廓清楚的巢,被大量丰富的纤维或纤维粘液样的间质所包绕、肿瘤细胞岛的形状和体积不同,可以是极小的簇,也可以形成较大的巢或束、在大的细胞巢中常能见到中央坏死灶和出血(图1),粘液变性和钙化也是常见的特点、有时囊腔结构被上皮样细胞围绕,易误认为癌或上皮样间皮瘤,常能见到肿瘤细胞围绕间质呈栅栏状排列,部分病例可见到菊形团,高倍镜见肿瘤细胞紧密集聚在一起,胞膜不清楚、细胞呈未分化状,小而圆或卵圆形,细胞核染色质浓,但有时可出现泡状核,核仁不清楚,胞浆极少、多数病例肿瘤细胞比较均一,核的形状比较规则(图 2,3)、约 1/ 3的病例细胞核有异型性,个别可出现多形性和非典型性(图4)、核分裂常能找到,多数为>2个/HPF、通常肿瘤的间质成分与肿瘤细胞团的面积大致相等。但有时一些肿瘤富于细胞,而另一些肿瘤以间质成分为主。如果从这样一些区域取到很少的标本,或者细针穿刺,往往很难做出DSRCT的诊断、间质一般由致密的胶原束组成,内含一致的纤维母细胞样或肌纤维母细胞样的梭形细胞,这些细胞平行排列在肿瘤细胞巢的周围,包绕单个细胞巢。间质中粘液样变和玻璃样变常能见到、个别病例基质细胞也能见到核分裂。
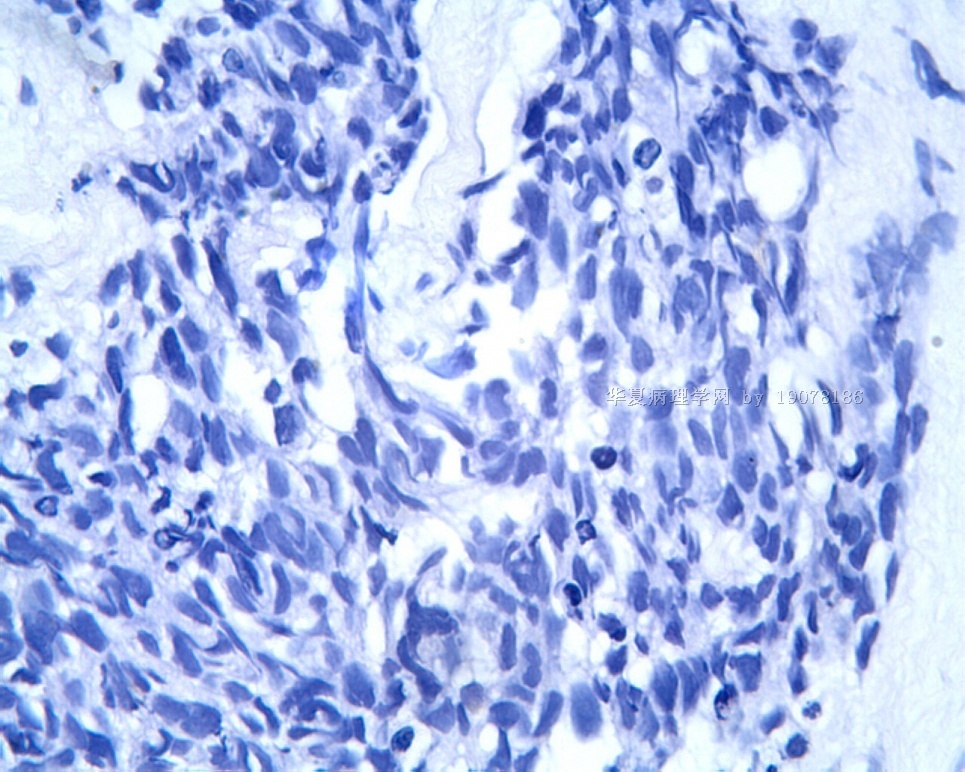
3.2 DSRCT的其它组织学类型 除了上述典型的组织学改变外,约有1/3的DSRCT还可以出现其它的组织形态,当以这些变化的组织形态为主时,常被误诊、这些形态包括:①梭形细胞结构:有的病例在肿瘤的大部分区域中细胞呈梭形,也可出现灶状梭形细胞区(图5)、至少有半数病例在光镜下虽然见不到梭形细胞,但经免疫组化染色后就可见到明显的梭形细胞、另外,个别病例在接受化疗后其复发的肿瘤出现梭形细胞形态。②类癌样结构:肿瘤主要由弥漫增生的小细胞组成,肿瘤细胞可排列成Homer- Wrinht样菊形团结构,个别病例可以没有显著的硬化,肿瘤细胞巢被纤维血管束分隔(图3,6)。③横纹肌样结构:肿瘤细胞胞浆有时呈轻度嗜酸性,但PAS染色阴性,胞浆内嗜酸性包含物使核受压,呈横纹肌样(图7)、④小管样结构:最近报道的一组材料中有8/39的病例在肿瘤细胞巢中出现小管样形态,尤其是在多集性肿瘤中比较显著,肿瘤细胞沿着被覆扁平或立方细胞的腔隙生长,这些集腔一般呈无规则形。⑤腺囊样结构:个别病例的部分区域形成假菊形团样或腺囊样,高倍镜见假菊形团,中央和腺囊样腔内为粘液样基质,易误诊为腺样囊腺癌。③透明细胞样结构:个别区域肿瘤细胞巢是由透明细胞组成,胞浆界限相对清楚(图8)。①印戒细胞样:细胞有透明胞浆,核偏位,呈印戒细胞样(图9)。②器官样结构:在一些实性区域中上皮样的肿瘤细胞较明显,井排列成器官样,产生细胞球(zellenballen)样形态。②移行细胞癌样结构:个别肿瘤的某些区域的细胞巢非常像移行细胞癌(图10),另外,一例报道肿瘤的某些区域细胞呈单行、背靠背的列兵样排列,似浸润性小叶癌,在一个肿瘤中可以同时出现上述凡种不同的形态,也可以以某一形态为主,但一般都或多或少的有典型结构存在。
4 兔疫组织化学
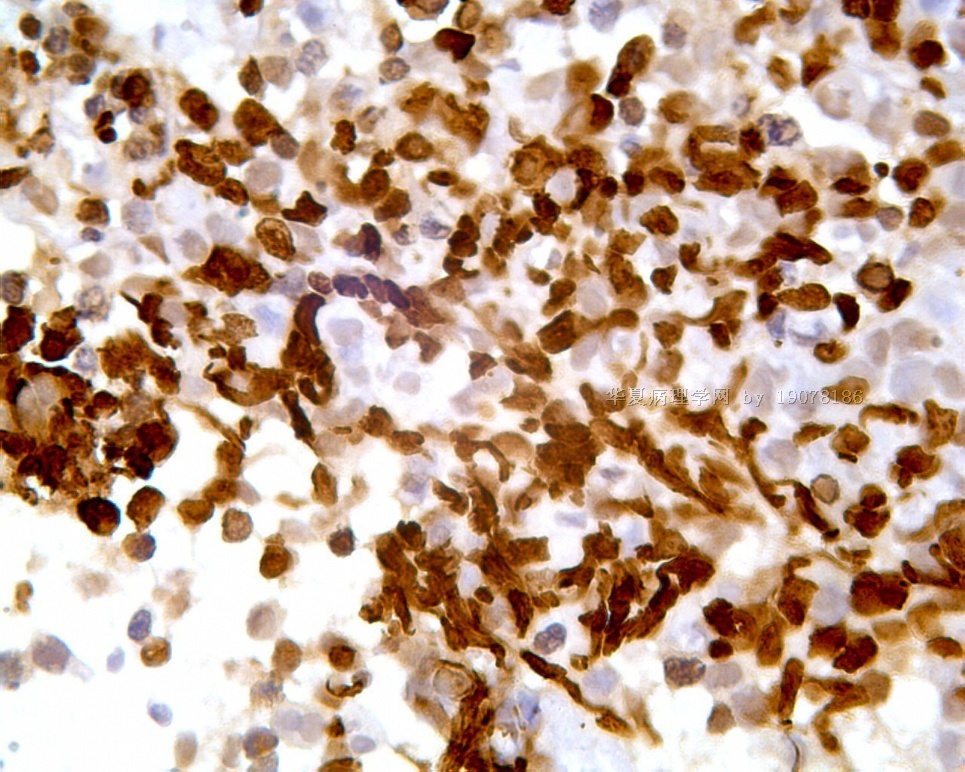
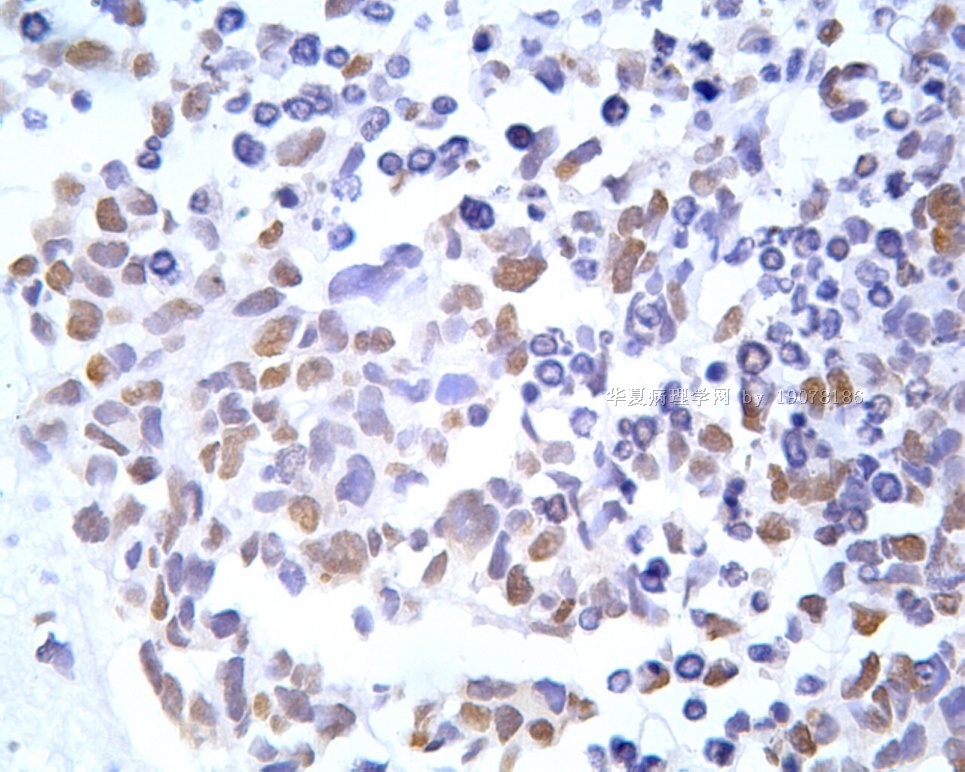
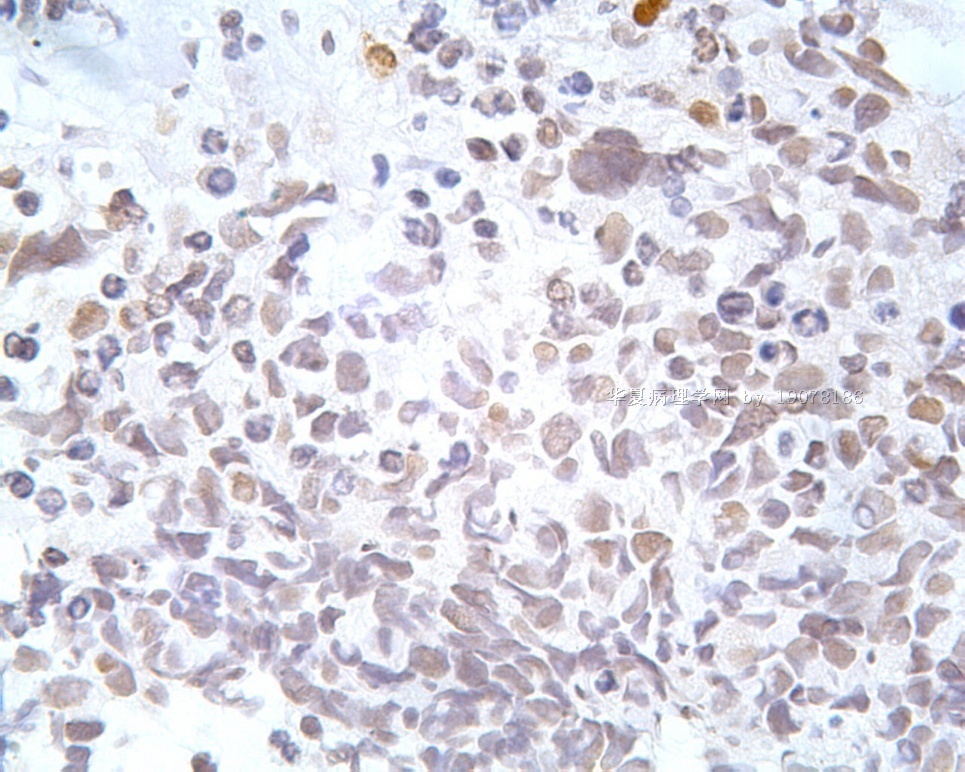
凡乎所有病例都表达desmin,cytokeratin和Vimentin。约3/4的肿瘤对 desmin的表达强而弥漫,并且近75%的病例阳性物质靠近核呈小球样。Vimentin表达一般无规律,占80%以上的病例有阳性反应,部分病例也可出现小球样结构。Cytokeratin的表达为复合性的,对AE1/AE3和CAM5.2抗体均可出现反应,尤其是采用"鸡尾酒”法对这三种抗体进行同时染色,阳性率可达 95%,多数病例为弥漫性强阳性反应。但对cytokeratin 5/6和 cytokeratin 20不表达,EMA的阳性率很高,一般位于胞浆中,并且个别cytokeratin阴性者对EMA可出现阳性反应,其它上皮性标志物也常表达,如MOC-31、Ber-EP4和CA-125、MOC-31是一种从肺小细胞癌细胞株克隆出来的识别 38 KD上皮糖蛋白的单克隆抗体,表达位于细胞膜上,且较强。Ber-EP4抗体来源于MDF-7乳腺癌细胞株的膜片段,大约有71%的 DSRCT对此抗体发生阳性反应。CA125是一种高分了量糖蛋白,常存在于卵巢粘液癌和内膜腺癌中,但部分DSRCT可对它表达,WT1是肾母细胞瘤位于染色体11的基因,DSRCT凡乎均对多克隆抗体WT1蛋白抗体有阳性反应,少数病例对CD99有表达,CD99是一个被位于X和Y性染色体假启动子基因编码的30-32KDa细胞表面的糖蛋白,绝大多数尤文氏肉瘤和PNET对此发生反应,有半数以上的肿瘤表达NSE和CD57(Leu-7),少数病例表达触突素和 CgA、仅有几例肿瘤表达肌特异性actin(MSA和a-平滑肌 actin(a-SMA)、其它抗体如 Sl00蛋白、HMB45、GFAP和NF等均为阴性。
5 超微结构特征
通常电镜下可以见到肿瘤细胞显示未分化性,细胞紧密连在一起,细胞器少,在肿瘤细胞巢和基质之间可见薄的基膜、细胞核的形状不同,有时不规则、染色质分散,但时而积聚成团块状、核仁一般见不到、大多数肿瘤细胞的连接不丰富,常常分化较差、少数肿瘤可以见到小的较成熟的带张力细丝的桥粒,个别肿瘤这种桥粒很多、一些肿瘤细胞可以见到少量的糖原,也常见到较丰富的自由核糖体和短的粗面内质网,以及中等量的线粒体、部分病例胞浆内含有脂滴。有时肿瘤出现含微管的树枝样突起、在具有神经标志物反应的肿瘤中可以见到致密核心颗粒,个别肿瘤有稀少的神经内分泌样颗粒、少数病例在细胞间或细胞内形成囊腔,囊腔表面有短的微绒毛、光镜所见到的印戒样细胞区,其电镜显示为胞浆内空泡形成。
在大多数病例中,一个最常见的现象是在核旁的胞浆中存在积聚的中间丝,这种中间丝在不同的病例或同一肿瘤的不同部位之间变化较大、有些细胞仅有小的积聚物,而另一些细胞中积聚物很大,可占据大部分胞浆,并且常常排列成球形涡轮状,与免疫组化中的球状物相对应、这些中间丝的积聚物不仅在圆形细胞中可见到,在梭形细胞中亦能见到、在所研究的病例中,没有找到能够提示向平滑肌和骨骼肌分化的具有密体和Z带样物质的中间丝。
基质细胞为典型的纤维母细胞或肌纤维母细胞,后者可见到粗面内质网和带有密体的长细丝束。
6 细胞遗传学特征
通过基因分析研究,几乎所有病例在染色体 t(11:222)(pl3;qll或q12)都有异常,这是该肿瘤一个特殊的表现。在第22号染色体上影响DSRCT的区域与尤文氏肉瘤基因(EWS)十分密切,EWS在尤文氏肿瘤/PNET(t11:222)(p24:q12)、透明细胞肉瘤(t(12:22)(q13:q12)和粘液软骨肉瘤(t (9:22)(q22-31;q12))中为特异性的肿瘤易位基因。尽管这些肿瘤的基因断裂点不完全相同,但都在染色体 22q12区。22ql2条带位于EWS基因位点,此位点在这些肿瘤中都被重排、而在第11号染色体上的损坏点是肾母细胞瘤的抑制基因(1l p13)。最近有人研究26例DSRCT,其中25例被证明有EWS-WT1基因融合,其基因损坏点在WT1第7内显子和EWS-WT1的7、8、9内显了上。因此,DSRCT可能是染色体区2个断裂点的结合,而这2个染色体区域在其它儿童的小细胞肿瘤中具有重要意义。该肿瘤的异向分化可能是这种基因重组变化的作用、肾母细胞瘤多向分化的能力己经众所周知,此肿瘤的母细胞成分能够表达Vimentin和Cytokeratin,并且部分病例也表达NSE,此外基质成分明显地向肌源性分化、尤文氏瘤/PNET可以向神经分化,这种分化与预后差有关。EWS与WT1基因的配对决定了肿瘤的表犁.其结果使DSRCT结合了尤文氏肉瘤/ PNET向神经分化和肾母细胞瘤多向分化(尤其是上皮分化)的特点。
7 鉴别诊断
鉴别诊断应包括各种发生于儿童和年轻患者的小细胞性肿瘤:横纹肌肉瘤、尤文氏肉瘤/PNET、神经母细胞瘤、肾母细胞瘤、胶质瘤、单形性滑膜肉瘤、小细胞性间皮瘤和非霍奇伞淋巴瘤等、另外,当病变不是以典型形态出现,而是以其它组织类型为突出表现时,也要与相应的肿瘤鉴别,如横纹肌样瘤、转移性腺癌、肉瘤样癌和其它的梭形细胞肉瘤等。
患者的年龄和发生部位(腹部)是鉴别诊断的一个重要因素、例如,肾母细胞瘤和神经母细胞瘤很少发生在18岁以上的患者,而间皮瘤又很少发生在儿章和青少年,尤其是小细胞间皮瘤必须是老年患者才考虑;象滑膜肉瘤一般发生在四肢、此外,鉴别诊断主要依据肿瘤的典型组织形态、免疫表型和基因的变化进行鉴别,即:形成巢状的小细胞和硬化性间质,向上皮、间叶和神经分化的免疫表型,以及EWS和WT1融合的基因变化、还有几个方面可用于鉴别:①横纹肌肉瘤:要着重与腺泡状横纹肌肉瘤鉴别、除了DSRCT有多向分化的特点外,虽然两者都表达desmin,但DSRCT很少表达SMA和MSA,且电镜找不到向肌分化的证据。新近发现的MyoD1和myogenin等是肌源性基因的调节因子,也称基因调节蛋白,它们在骨骼肌分化程序中的表达比 myosin,actint和 desmin等结构蛋白早得多,抗MyoD1和 Myogenin的抗体己应用于分化差的横纹肌肉瘤的诊断,而这两种抗体被一些学者证明在DSCRT中不表达、因此有人认为desmin在DSRCT中的表达不能表明有骨骼肌分化、②尤文氏肉瘤/PNET:与DSRCT比较,不但在形态上而且在免疫表型和基因型上都有很多的相似之处,而且都可有神经标记物和CD99表达,以及EWS基因存在。DSRCT表达显示神经分化的标记物有NSE和CD57,但不表达NF和GFAP;而尤文氏肉瘤/PNET可以表达NF和GFAP、DSRCT表达CD99为灶状散在;后者一般为弥漫性。另外,DSRCT表达WT1蛋白,而后者不表达此蛋白。DSRCT大多数desmin阳性,其阳性染色在胞浆内呈圆球状是一个特点,尤文氏肉瘤/PNET则罕见、③肾母细胞瘤:腹膜后发生的DSRCT有时要与此肿瘤鉴别,除了上述所提到的形态特点外,肾母细胞瘤表达WT1而不表达EWS。④转移性癌:一般不难与DSRCT鉴别、对于有印戒细胞样结构的DSRCT,一个简单的鉴别法就是进行粘液染色,因为DSRCT的印戒样结构不是由细胞产生粘液造成的,而是细胞内腔的形成、高度扩张的粗面内质网或脂滴的存在所构成。
8组织起源
DSRCT的组织起源仍不清楚,但是文献中提出了一些不同的假设。由于该肿瘤的分化水平较低,且主要发生在儿章和青年人,所以有人认为可能来自于具有多向分化潜能的、胚胎或胎儿前细胞恶性转化的结果。有神经抗原的表达和可形成菊形团的结构,使一些研究者认为此肿瘤是神经外胚层起源,另外,多数病例发生在腹部,沿着浆膜面呈结节性扩散,引起人们考虑间皮起源的可能性,并且这种可能性符合肿瘤细胞的上皮和间质双向分化的特点、这种复合性的表达在胎儿的浆膜和成人的间皮瘤中均存在。目前,由于基因水平的研究结果,使后一种认识又有进一步的深入,即WT1和EWS基因的融合,从而使肿瘤结合了尤文氏肿瘤和肾母细胞瘤的特点,向多方向分化、WT1基因蛋白不仅在肾母细胞瘤和DSRCT中表达,也能在泌尿生殖管道、胎儿腹和胸膜间皮表达,而且在一些间皮瘤中也能检测到,并且,有人认为WT1基因可引起间皮向上皮分化、有人还提出间皮下组织是从相同的内脏膜的内胚层组织分化而来,这种内脏膜的内胚层组织也能形成肾赃,这些均支持肿瘤来源于间皮或间皮下组织。
9预后与治疗
文献中报道的绝大多数病例肿瘤呈进行性生长,患者预后不好、一组有35例随访的报道,其中25例于诊断后8- 50个月死亡(平均 25 2个月),均死于肿瘤广泛转移、另外10例带瘤生存,最长仅31个月。肿瘤对放疗不十分敏感,中于腹腔内器官对放疗的低耐受性和肿瘤的多发性,不易采用放射疗法、手术后常采用化疗,大多数病例最初肿瘤体积缩小,一部分患者可对化疗产生积极地反应、有人报道了17例接受化疗的患者,有8例达到完全反应状态,其中3例复发,另外5例肿瘤完全消失,持续时间4-48个月、但最终的生存率令人失望,患者常常又出现肿瘤进行性生长、治疗仍是急待解诀的问题。

- 一念天堂,一念地狱。
-
本帖最后由 于 2010-05-27 21:45:00 编辑
CK没做,正打算做CK、EMA,结果可能于星期二出来。暂倾向于考虑:恶性蝾螈瘤。
Malignant triton tumor (MTT) is a relatively rare, aggressive tumor comprised of bothmalignant schwannoma cells and malignant rhabdomyoblasts. Malignant peripheral nerve
sheath tumors (MPNSTs) are neoplasms derived from the cellular constituents of the peripheral nerve sheath. The majority arise from Schwann cells,1but some could develop from fibroblasts and supporting cells known as perineural cells. On occasion, there are focal histological variants of epithelial, glandular,or mesenchymal elements. This divergent differentiation occurs only in 15–20% of all MPNST cases2.Rhabdomyosarcomatous elements are the most common within MPNSTs, constituting a
subtype—the malignant triton tumor (MTT). Tumors with osteosarcomatous or chondrosarcomatous differentiation are rarer,reported in approximately 5% of all MPNST cases, while they are rarely seen in MTTs.
- 一念天堂,一念地狱。